转录专题

Easy Voice Toolkit - 简易语音工具箱,一款强大的语音识别、转录、转换工具 本地一键整合包下载
Easy Voice Toolkit 是一个基于开源语音项目实现的简易语音工具箱,提供了包括语音模型训练在内的多种自动化音频工具,集成了GUI,无需配置,解压即用。 工具箱包括 audio-slicer、VoiceprintRecognition、whisper、SRT - to - CSV - and - audio - split、vits 和 GPT - SoVITS 等。这些优秀

杨树84K品种的单细胞测序发现转录因子PagMYB31的功能-文献精读44
Transcription factor PagMYB31 positively regulates cambium activity and negatively regulates xylem development in poplar 转录因子PagMYB31正向调控杨树84K品种的形成层活动,并负向调控木质部的发育。 同样有篇文献,二倍体毛白杨基因组~ 二倍体毛白杨(Populus

零基础入门转录组数据分析——单基因ROC分析
零基础入门转录组数据分析——单基因ROC分析 目录 零基础入门转录组数据分析——单基因ROC分析1. ROC分析的基础知识2. 单基因ROC分析(Rstudio)——代码实操2. 1 数据处理2. 2 单基因ROC分析2. 3 ROC曲线简单可视化 1. ROC分析的基础知识 1.1 ROC分析是什么? ROC(Receiver Operating Characte
零基础入门转录组数据分析——预后模型的验证
零基础入门转录组数据分析——预后模型的验证 目录 零基础入门转录组数据分析——预后模型的验证1. 预后模型的基础知识2. 预后模型的验证(Rstudio)——代码实操2. 1 数据处理2. 2 构建多因素cox模型(用输入的全部5个基因)2. 3 计算风险评分2. 4 生存分析(验证一)2. 5 ROC分析(验证二) 1. 预后模型的基础知识 关于预后模型的其他基础

zoom 会议 javascript 转录例子
一、启动server-to-server zoom api服务,用于创建会议,参考:如何使用Zoom API创建一个会议?-CSDN博客 二、启动meetingsdk-auth-endpoint服务,用于加入会议,参考:zoom 会议机器人web例子-CSDN博客 三、修改meetingsdk-javascript代码 四、自带转录效果CC(但没有对外提供转录接口)

ChIP-seq项目文章 | Adv Sci转录因子FOXK1通过驱动小管上皮细胞糖酵解促进慢性肾脏疾病
在慢性肾脏疾病(CKD)进展过程中,肾小管上皮细胞(TECs)经历了从脂肪酸氧化到糖酵解的能量相关代谢转变。然而,这种糖酵解爆发的机制尚不清楚。2024年7月31日,武汉大学王惠明教授团队和湖北民族大学刘伦志教授团队在Advanced Science(IF:14.6)上在线发表了题为“Forkhead Box Protein K1 Promotes Chronic Kidney Disease b
零基础入门转录组数据分析——预后模型之lasso模型
零基础入门转录组数据分析——预后模型之lasso模型 目录 零基础入门转录组数据分析——预后模型之lasso模型1. 预后模型和lasso模型基础知识2. lasso预后模型(Rstudio)——代码实操2. 1 数据处理2. 2 构建lasso预后模型2. 3 提取Lasso预后基因2. 4 计算风险评分 1. 预后模型和lasso模型基础知识 1.1 预后模型是

使用Mikado挑选最好的转录本进行注释
Mikado是基于Python3写的基因组结构注释工具,它主要做的是从多个转录组组装工具得到的转录本里挑选出最好的结果作为基因组的结构注释。此外,它还会基于同源蛋白比对结果对转录本打分。换句话说这个软件主要是根据转录组数据进行注释,没有 ab inito 预测。 软件安装比较方法,我们可以使用bioconda进行安装: conda create -n mikado mikado# 打开Pyt

「学转录组入门生信」第二周来获取表达量矩阵
我们第二周目标有四个: 整理数据RNA-seq格式了解数据质控数据比对read定量 首先,我们得要知道我们在转录组分析过程中会遇到很多格式,建议先通过搜索查找了解这些格式是什么 fasta/fas/fagtf/gffbedsam/bamcsv/tsv/txt 接着,我们会在分析过程中时刻检查我们的数据质量,所以你要尝试回答下面这几个问题 数据质控要在哪个阶段做不同阶段要看什么标准质控有哪

「学转录组入门生信」第一周从环境配置开始
image 我们第一周目标有三个: 熟悉Linux环境 登录服务器Linux基本命令PATH的意义学习conda管理环境 如何在conda中添加channel如何用conda安装和卸载软件如何创建新的环境和切换环境数据准备 首先,你需要有一个Linux环境,Windows10用户可以安装WSL,MacOS请在应用程序中搜索终端 Windows10配置WSL: https
「单细胞转录组系列」如何从稀疏矩阵中提取部分数据进行分析
这一篇文章是回答知识星球中一位星友的提问,她的电脑内存有限,无法直接使用所有数据,只能分析部分数据。 数据来源: https://content.cruk.cam.ac.uk/jmlab/atlas_data.tar.gz 解压缩之后,得到下面数据 数据清单 其中raw_counts.mtx是以稀疏矩阵格式存放的表达量数据,文件为6.5G, 用普通的文本编辑器无法打开,
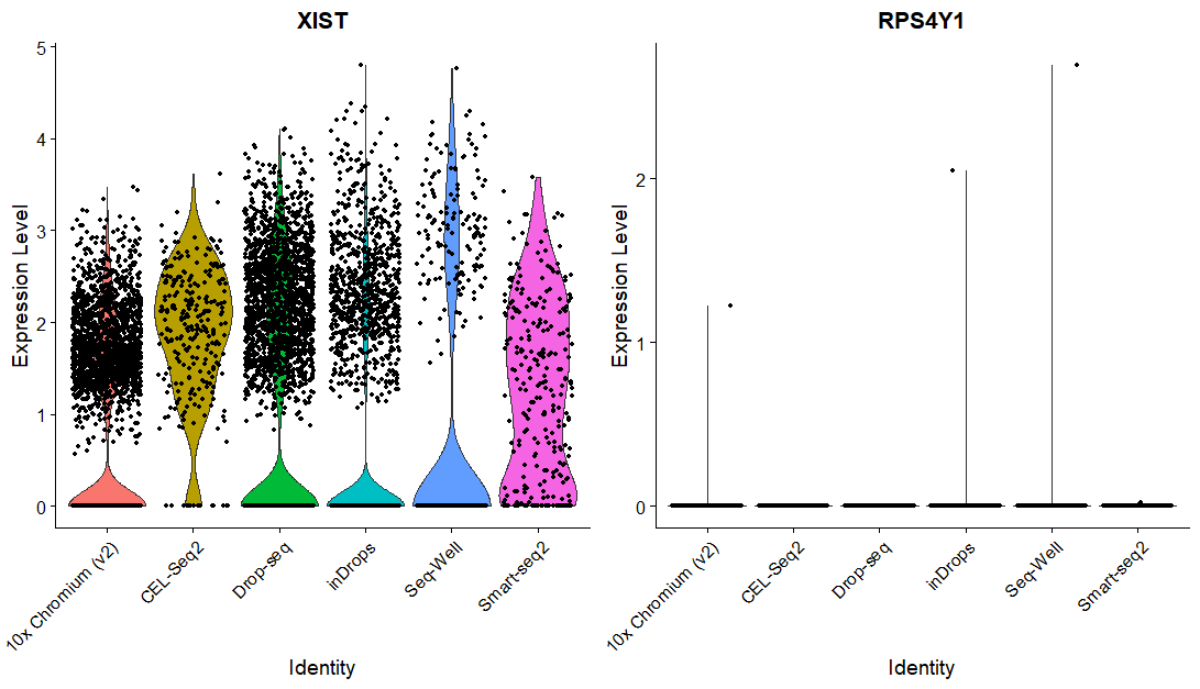
如何确定公共转录组数据集的来源性别
太长不看版: 文献报道XIST和RPS4Y1是区分性别的两个高可信度的标记基因,因此你没有必要去用其他性染色体上的基因去确定数据集的性别。 不仅仅是在使用公共的单细胞转录组数据,其实早在公共芯片数据或者RNA-seq数据挖掘中,就有人在考虑一个问题,这个数据的元信息作者会不会搞错了呢? 以性别为例,我们很容易想到表达Y染色体上基因数据肯定是男性,但是我们也知道基因也不是任何时刻都表达,所以如
「单细胞转录组系列」如何可靠地确定公共数据集的性别
太长不看版: 文献报道XIST和RPS4Y1是区分性别的两个高可信度的标记基因,因此你没有必要去用其他性染色体上的基因去确定数据集的性别。 不仅仅是在使用公共的单细胞转录组数据,其实早在公共芯片数据或者RNA-seq数据挖掘中,就有人在考虑一个问题,这个数据的元信息作者会不会搞错了呢? 以性别为例,我们很容易想到表达Y染色体上基因数据肯定是男性,但是我们也知道基因也不是任何时刻都表达,所以如

不用写一行代码,deepseek结合腾讯云语音识别来批量转录Mp3音频
首先,打开window系统中的cmd命令行工具,或者powershell,安装腾讯云tencentcloud的Python库 pip install -i https://mirrors.tencent.com/pypi/simple/ --upgrade tencentcloud-sdk-python 然后,开通腾讯云的对象存储COS服务, 把要转录成文本的mp3音频文件上
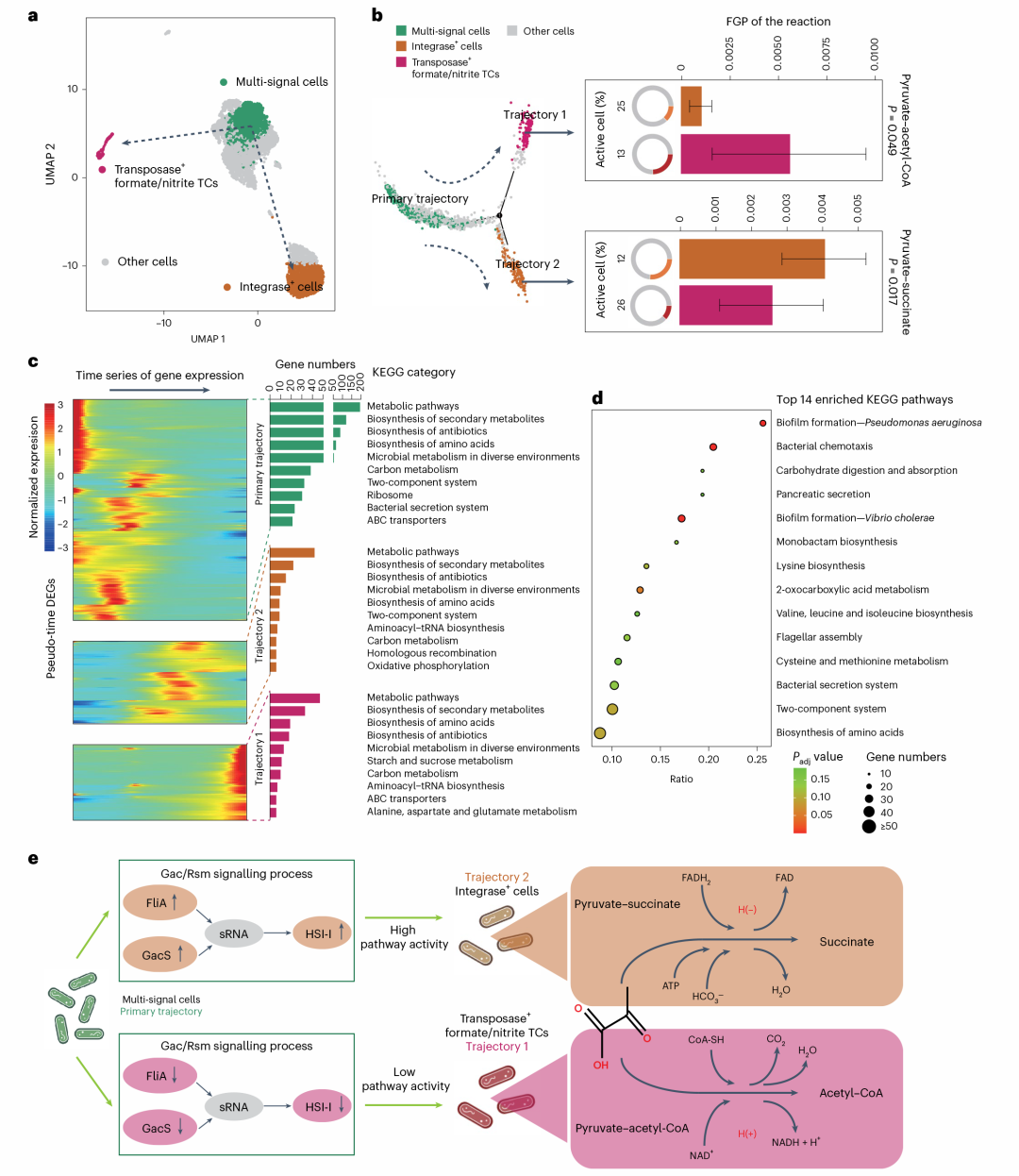
Nature Microbiology丨VITA单细菌转录组测序技术助力深入解析奶牛瘤胃微生物组功能异质性
瘤胃微生物组一直以来都是研究相对不足但又极其复杂的微生物生态系统之一。瘤胃微生物能够有效降解植物纤维,将其转化为高质量的蛋白质产品,在这一过程中,由于微生物强烈的发酵,还会产生大量气体,其成分主要包括二氧化碳和甲烷等温室气体,还有少量的氮气和微量的氢气、氧气和硫化氢,这些排放的温室气体甚至会对全球环境造成较大的影响。近些年以来,在宏组学技术的推动下,对瘤胃微生态系统功能的认识已取得了显著进展。然而
如何快速从基因组中提取基因、转录本、蛋白、启动子、非编码序列?
有读者留言想要提取外显子,内含子,启动子,基因体,非编码区,编码区,TSS上游1500,TSS下游500的序列。下面我们就来示范如何提取这些序列。 NGS基础 - 参考基因组和基因注释文件提到了如何下载对应的基因组序列和基因注释文件。 假如我们已经拿到了基因组序列文件GRCh38.fa和基因注释文件GRCh38.gtf,也可从文后链接获取。 查看下文件内容和格式 基因组序列文件为FASTA
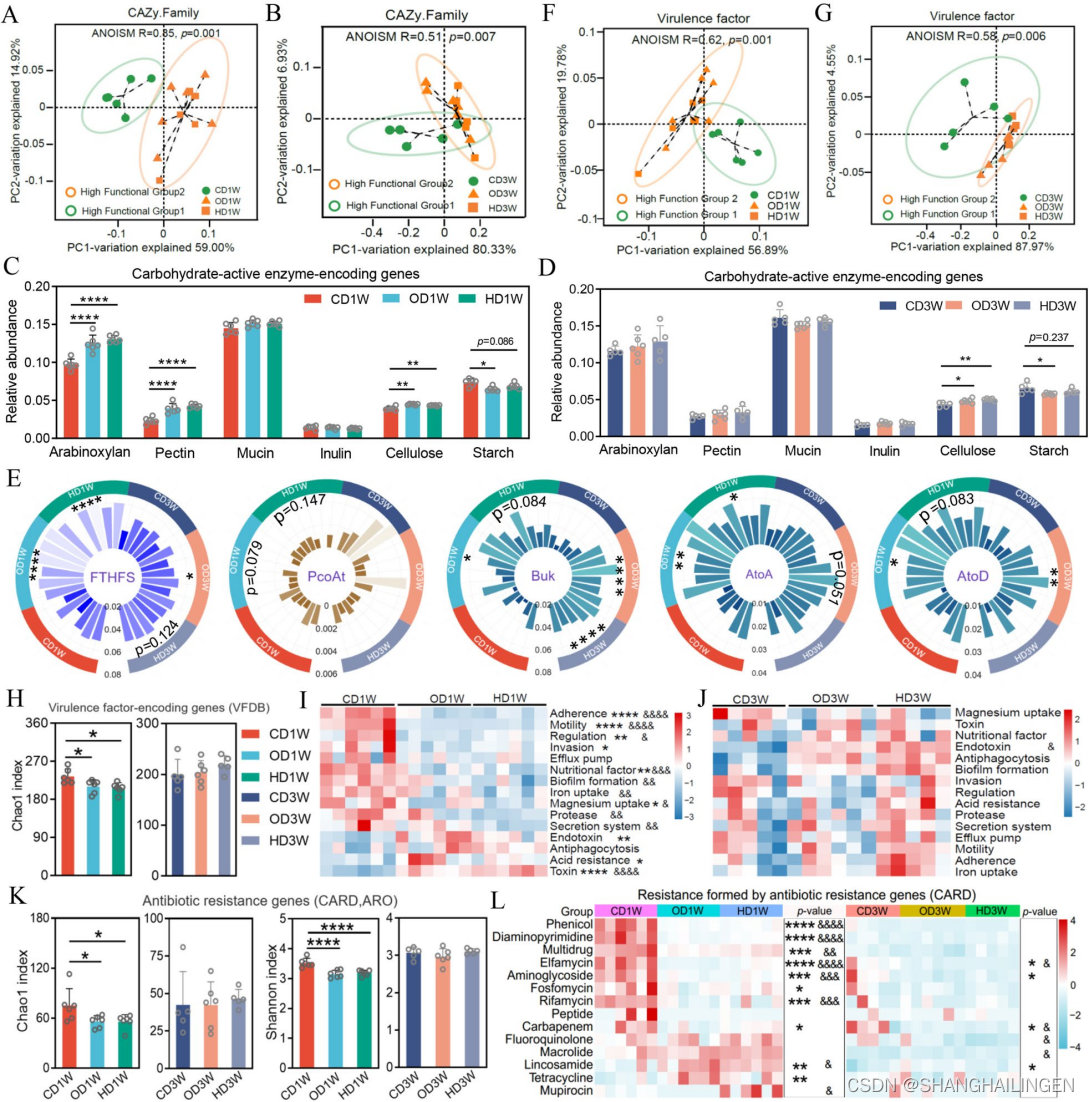
Microbiome | binning+转录组→首个草鱼肠道基因集目录发布啦
草鱼便宜又好吃 但是你了解草鱼吗? 草鱼的肠道里定殖着成千上万的共生微生物,它们与草鱼共同生存,相互影响。这些微生物在草鱼的新陈代谢、免疫调节等方面发挥着重要作用。 虽然同为经济作物,鱼类的微生态相关研究远远不如于其他畜禽,经济鱼类的微生物基因目录也尚未构建。 近期,来自中国农业科学院饲料研究所的研究团队,在《Microbiome》上发表了题目为《Decipher
Seurat | 不同单细胞转录组的整合方法
一、涉及的新概念 参考(reference):将跨个体,跨技术,跨模式产生的不同的单细胞数据整合后的数据集 。也就是将不同来源的数据集组合到同一空间(reference)中。 从广义上讲,在概念上类似于基因组DNA序列的参考装配。 查询(query):单个实验产生的数据集 转化学习(transfer learning):产生一个于参考数据集(reference)上进行训练的模型,可以将信
空间转录组基础数据解读+学习方法
详情请参考这个视频:空间转录组(spatial transcriptome)数据分析基础教程_哔哩哔哩_bilibili 1.首先是filtered_feature_bc_matrix文件 两个里面的内容本质一样,都是空间转录组 表达矩阵的信息 2.具体的所有东西可以在10x的网站里学,看参数 具体的网址:Cell Ranger Gene Expression Outputs -

专业、安全和快速及时的电话会议转录服务
在线搜索拨入式电话会议转录服务时,您会发现,无数“价廉物美”的DIY选择映入眼帘。但如果质量与成本一样重要,那么便需要一位经验丰富的专业转录提供商。 录制技术和语音文字转换软件的发展意味着,如果您想为重要电话会议创建一份书面记录,有很多DIY解决方案可供选择。 但在开始前,先问问自己为什么要录制和转录电话会议。如果会议重要到需要一份书面记录,那么这份记录是不是应该尽可能地准确、快速和安全? 电话会
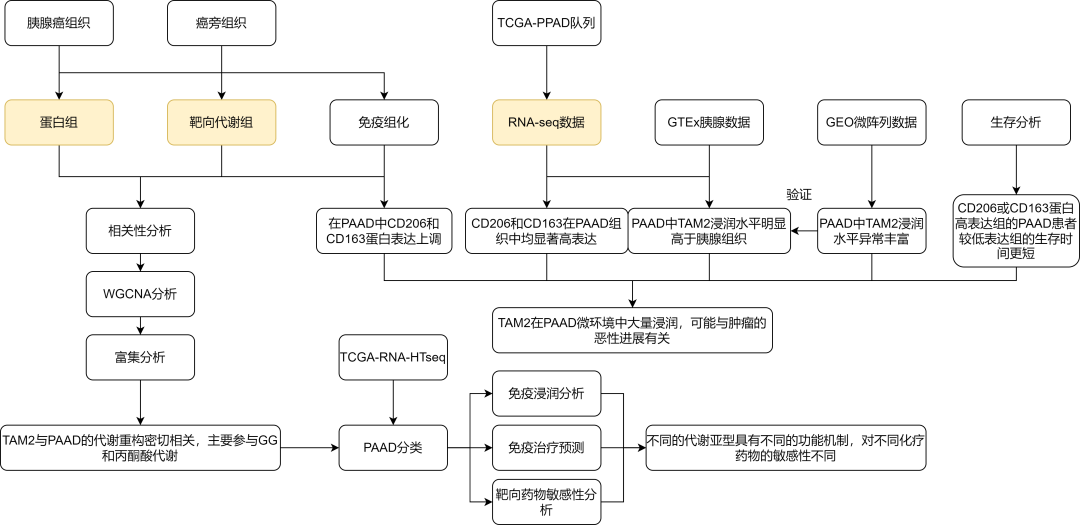
成功案例(IF=7.3)| 转录组+蛋白质组+代谢组联合分析分析揭示胰腺癌中TAM2相关的糖酵解和丙酮酸代谢重构
研究背景 肿瘤的进展和发展需要癌细胞的代谢重编程,癌细胞能量代谢模式的改变可以满足快速增殖和适应肿瘤微环境的需要。肿瘤微环境(TME)中的代谢状态受到多种因素的影响,包括血管生成、与其他细胞的相互作用和系统代谢。代谢异质性可影响治疗效果,并可能预测临床结果。然而,很少有研究关注基于代谢异质性来区分亚型的方法。 胰腺癌(PAAD)是一种预后不良、死亡率高的恶性肿瘤。肿瘤相关巨噬细胞(TA

数据分析:基于DESeq2的转录组功能富集分析
介绍 DESeq2常用于识别差异基因,它主要使用了标准化因子标准化数据,再根据广义线性模型判别组间差异(组间残差是否显著判断)。在获取差异基因结果后,我们可以进行下一步的富集分析,常用方法有基于在线网站DAVID以及脚本处理的两类,本文介绍基于fgsea的方法计算富集分析得分。 DESeq2差异分析 了解DESeq2如何标准化数据和识别差异基因。下面给出简要代码 library(DESeq
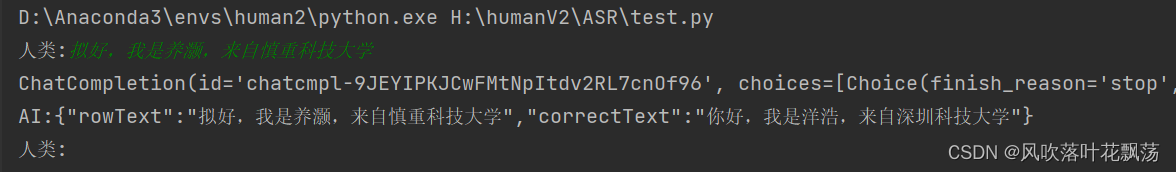
ASR语音转录Prompt优化
ASR语音转录Prompt优化 一、前言 在ASR转录的时候,我们能很明显的感受到有时候语音识别不是很准确,这过程中常见的文本错误主要可以归纳为以下几类: 同音错误(Homophone Errors) 同音错误发生在不同词语发音相似或相同的情况下。ASR系统可能难以区分这些词语的具体含义,从而导致错误的词语被识别。例如,中文里的“海”和“还”在某些方言或口音中发音相近,可能会被错误地互换。
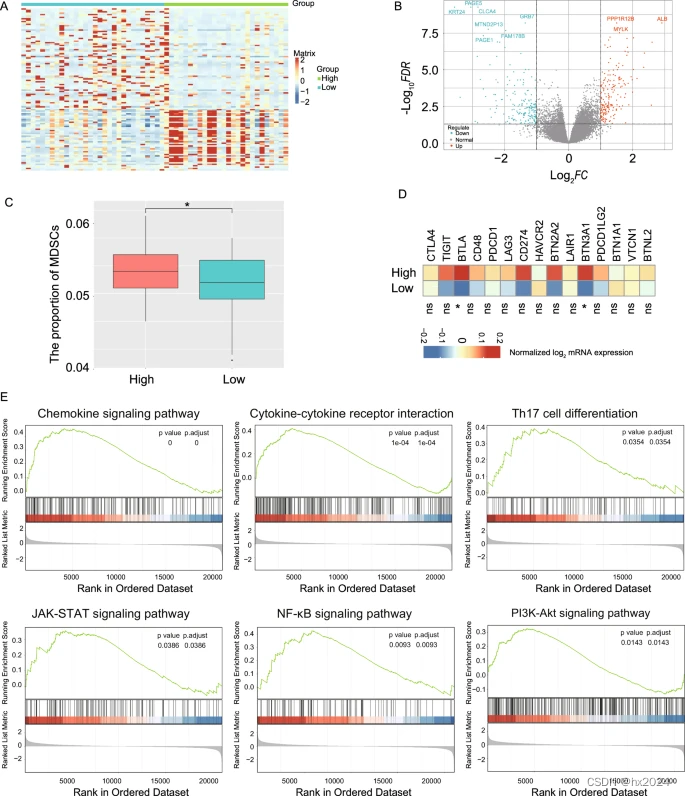
病理组学+配对 mIHC 验证+转录组多组学
目录 病理DeepRisk网络模型构建 DPS和新辅助化疗 mIHC 验证 STAD转录组层面 病理DeepRisk网络模型构建 自有数据训练,TCGA数据进行验证,然后配对mIF验证,最后还在转录组层面分析。 该模型基于中山数据集(n = 1120)构建,并在TCGA-STAD(n = 268)和SOBC数据集(n = 277)中得到进一步验证。 用于构建 DeepRisk

如何获取目标基因的转录因子(下)——Linux命令获取目标基因TF
如何获取目标基因的转录因子(上)一文中我们以人类基因组为例,从ensemble网站下载了基因组中基因位置信息矩阵GRCh38.gene.bed和基因组中转录因子结合位点信息矩阵GRCh38.TFmotif_binding.bed) 我们知道有很多数据库可以查找启动子、UTR、TSS等区域以及预测转录因子结合位点,但是怎么用Linux命令处理基因信息文件来得到关注基因的启动子和启动子区结合的TF呢

学习笔记Day21:转录组差异分析
转录组差异分析 差异分析难点在于将数据处理成需要的格式 表达矩阵 数值型矩阵-count 行名是symbol 低表达量的基因需要过滤 分组信息 因子,对照组在level第一位 与表达矩阵的列一一对应 项目名称 字符串(不要有特殊字符) TCGA-XXX 非TCGA数据特殊无要求 拿不到count数据如何做差异分析? 自行做上游分析得到count tpm:取log,用