生信专题

生信代码入门:从零开始掌握生物信息学编程技能
少走弯路,高效分析;了解生信云,访问 【生信圆桌x生信专用云服务器】 : www.tebteb.cc 介绍 生物信息学是一个高度跨学科的领域,结合了生物学、计算机科学和统计学。随着高通量测序技术的发展,海量的生物数据需要通过编程来进行处理和分析。因此,掌握生信编程技能,成为每一个生物信息学研究者的必备能力。 生信代码入门,旨在帮助初学者从零开始学习生物信息学中的编程基础。通过学习常用

生信圆桌x生信分析平台:助力生物信息学研究的综合工具
介绍 少走弯路,高效分析;了解生信云,访问 【生信圆桌x生信专用云服务器】 : www.tebteb.cc 生物信息学的迅速发展催生了众多生信分析平台,这些平台通过集成各种生物信息学工具和算法,极大地简化了数据处理和分析流程,使研究人员能够更高效地从海量生物数据中提取有价值的信息。这些平台通常具备友好的用户界面和强大的计算能力,支持不同类型的生物数据分析,如基因组、转录组、蛋白质组等。

生信软件34 - 大幅提升Python程序执行效率的工具Pypy
在生信开发过程中,会大量使用Python脚本,除了多进程和多线程编程提高程序运行效率外,还可以借助效率更高的Python解释器来提高程序的运行速度, CPython 使用c语言实现的解释器, PyPy 使用python语言的子集RPython实现的解释器,一般情况下PyPy比CPython快4倍左右。 1. 比较解释器的运行效率 # task.pyimport timedef compute

外泌体相关基因肝癌临床模型预测——2-3分纯生信文章复现——5.拷贝数变异及突变图谱(2)
内容如下: 1.外泌体和肝癌TCGA数据下载 2.数据格式整理 3.差异表达基因筛选 4.预后相关外泌体基因确定 5.拷贝数变异及突变图谱 6.外泌体基因功能注释 7.LASSO回归筛选外泌体预后模型 8.预后模型验证 9.预后模型鲁棒性分析 10.独立预后因素分析及与临床的相关性分析 11.列线图,ROC曲线,校准曲线,DCA曲线 12.外部数据集验证 13.外泌

生信机器学习入门3 - Scikit-Learn训练机器学习分类感知器
1. 在线读取iris数据集 import osimport pandas as pd# 下载try:s = 'https://archive.ics.uci.edu/ml/machine-learning-databases/iris/iris.data'print('From URL:', s)df = pd.read_csv(s,header=None,encoding='utf-8'

生信软件32 - 变异位点危害性评估预测工具合集
转换和颠换如发生在基因的蛋白编码区内,根据点突变对和蛋白质翻译产生的影响定性,可以把点突变分为同义突变和非同义突变二种。 1. 基本概念 1.1 同义突变(synonymous mutation) 碱基替换不引起氨基酸改变称为同义突变。 1.2 非同义突变(non-synonymous mutation) 碱基替换引起编码氨基酸改变称为非同义突变。 1.2.1 错义突变 碱基替换引起

生信圆桌x生信宝典:掌握生物信息学的全面指南
少走弯路,高效分析;了解生信云,访问 【生信圆桌x生信专用云服务器】 : www.tebteb.cc 介绍 生物信息学作为跨学科的前沿领域,融合了生物学、计算机科学、数学和统计学等多种学科知识,已成为现代生物学研究中不可或缺的一部分。然而,由于其学科交叉性强、内容庞杂,许多初学者在进入这个领域时常感到困惑和迷茫。《生信宝典》正是为了解决这一问题而编写的,旨在为生物信息学的初学者和

生信圆桌:专业生信服务器与平台服务的提供者
生信圆桌是一个专注于提供生物信息学(生信)服务器和平台服务的领先企业,致力于为全球科研机构、企业和独立研究者提供高性能的生信分析解决方案。随着生物信息学研究对计算资源的需求日益增加,生信圆桌凭借其先进的服务器技术和专业的服务团队,成为了生信领域中不可或缺的合作伙伴。 访问生信圆桌,使用生信云。高效分析少走弯路 www.tebteb.cc 生信圆桌的核心服务 高性

生信圆桌x生信友好期刊:助力生物信息学研究的学术平台
介绍 生物信息学作为一门交叉学科,近年来得到了快速发展。为了促进生信领域的科研交流,许多学术期刊开始关注并专门发表生物信息学相关的研究成果。这些期刊被称为“生信友好期刊”,它们为研究人员提供了一个展示和传播最新科研成果的重要平台。 访问生信圆桌,使用生信云。高效分析少走弯路 www.tebteb.cc 生信友好期刊的主要特点 专注生信领域: 生信友好期刊专注于生物信

用Python实现生信分析——次结构预测详解
次结构预测是指预测生物大分子(如RNA和蛋白质)在不考虑其三维结构的情况下的局部折叠模式。次结构通常指二级结构,例如RNA中的碱基对或蛋白质中的α-螺旋和β-折叠。通过预测这些结构,我们可以更好地理解分子的功能和作用机制。 1. RNA二级结构预测 RNA二级结构 主要由碱基配对形成,包括发夹(hairpin)、茎环(stem-loop)、假结(pseudoknot)等结构。RNA二级结构预测

生信技能57 - Samtools获取指定外显子区域depth和提取BAM文件序列
1. Samtools depth 根据指定bed文件,获取指定区域的覆盖度信息。 # 提取IDT xGen V1 HBA1 exon bedcat xgen-exome-hyb-panel-v1-targets-hg19.bed|grep -w HBA1 > hba.exon.bed# 提取HBA1 外显子的覆盖度# -b: 提取depth的bed文件samtools depth -b

生信是什么?生物信息学的基础概念与应用领域-生信圆桌
介绍 生信,全称为生物信息学(Bioinformatics),是指将计算机科学、数学和统计学的方法应用于生物学数据的处理、分析和解释。随着基因组测序技术的发展和大规模生物数据的产生,生物信息学成为了生命科学研究中的一个核心领域。它通过整合和分析大量的生物数据,揭示基因组、蛋白质、代谢物等生物分子的复杂关系,从而推动医学、农业、环境科学等多个领域的进步。 生信的主要内容 生物信

生信圆桌x生信期刊整理:探索前沿生物信息学研究成果的重要学术平台
随着生物信息学领域的快速发展,越来越多的研究者开始将他们的研究成果发表在生物信息学期刊上。这些期刊不仅是学术界分享最新研究发现的重要平台,也为研究人员提供了展示和传播其研究工作的机会。生信期刊涵盖了从基因组学、转录组学、蛋白质组学到系统生物学、计算生物学等多个领域,是生物信息学领域的重要学术资源。 [生信圆桌]https://www.tebteb.cc 主要的生信期刊概览 以

生信圆桌x生信豆芽菜:生物信息学新手的学习与成长平台
生信豆芽菜是一个专门为生物信息学初学者创建的学习与交流平台,致力于帮助新手们快速入门并掌握生信分析的基础知识与技能。随着生物信息学在科研中的重要性日益提升,越来越多的学生和研究人员开始接触这一领域。生信豆芽菜正是为了满足这些新手的需求,提供了丰富的学习资源、实用的工具介绍和活跃的社区支持。 【生信圆桌】生信圆桌x生信专用云服务器 - 生信云,让生信分析变简单高效 生信豆芽菜
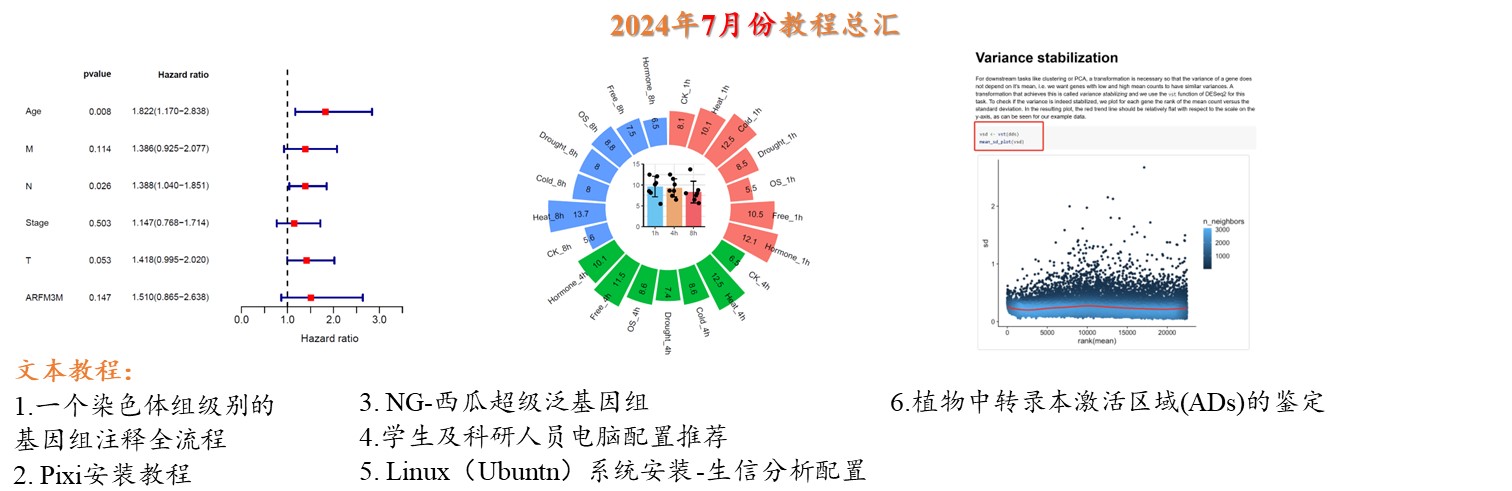
关于我的生信笔记开通《知识星球》
关于知识星球 1. 为什么到现在才开通《知识星球》 从很早关注我的同学应该了解小杜的知识分享历程,小杜是从2021年11月底开始进入此“坑”,一直坚持到现在,马上3年了(24年11月底到期)。自己也从一个小青年,变成一个大青年。也许后面,会一直更新下去,但是也许我某一天突然间就停止了(PS:事事难料啊)。 你说分享3年的时间长或是不长呢? 说长,对的。 谁又有几个3年的时间一直投入这件

可视化生信分析利器-Galaxy(第一讲)
什么是Galaxy 很多公司开始推广他们的可视化生信分析工具,有人说未来的趋势是无代码,分析只要拖拖点点就行了。无代码只能说是一个噱头,毕竟人人都会“用"excel,也不是人人都是数据分析师。 但是一个数据分析师肯定知道如何正确的使用excel,所以一个真正的生信媛/猿也不会嫌弃那些可视化的工具。毕竟写代码累了,没事拖拖点点也是别样的乐趣。 Galaxy就是很多年前在云计算背景下诞生的开源项
「学转录组入门生信」第二周来获取表达量矩阵
我们第二周目标有四个: 整理数据RNA-seq格式了解数据质控数据比对read定量 首先,我们得要知道我们在转录组分析过程中会遇到很多格式,建议先通过搜索查找了解这些格式是什么 fasta/fas/fagtf/gffbedsam/bamcsv/tsv/txt 接着,我们会在分析过程中时刻检查我们的数据质量,所以你要尝试回答下面这几个问题 数据质控要在哪个阶段做不同阶段要看什么标准质控有哪
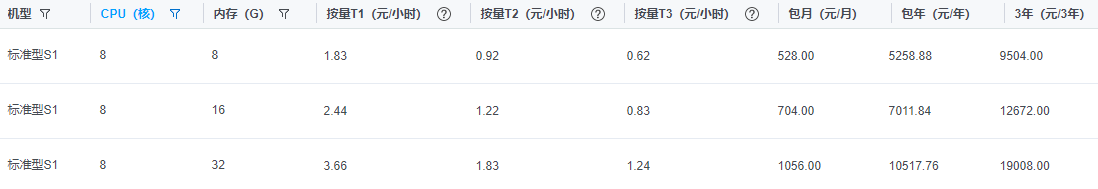
「生信基础课」如何利用好手头的电脑,节省上千的服务器租用费
对于一个专业的生信分析人员,一台高性能的服务器是必不可少的,因此会在上面投入能力承受范围的资金。 关于为何要用Linux系统,我录制了一个2分钟的视频,https://www.bilibili.com/video/av58133450 但是对于一个仅仅想了解生信是什么, 想跑跑简单的流程的初学者而言,动辄月租上千的服务器并不是好的选择。 某厂商报价 最好的策略是先

「学转录组入门生信」第一周从环境配置开始
image 我们第一周目标有三个: 熟悉Linux环境 登录服务器Linux基本命令PATH的意义学习conda管理环境 如何在conda中添加channel如何用conda安装和卸载软件如何创建新的环境和切换环境数据准备 首先,你需要有一个Linux环境,Windows10用户可以安装WSL,MacOS请在应用程序中搜索终端 Windows10配置WSL: https

「生信Debug」OpenBLAS blas_thread_init: pthread_create: Resource temporarily unavailable
BLAS(Basic Linear Algebra Subprograms),翻译为基础线性代数子程序库,里面拥有大量已经编写好的关于线性代数运算的程序。OpenBLAS是其中一个实现了相关运算的开源程序库,其他软件在开发的时候就不需要额外造轮子,直接调用相关的API即可。 之前在使用OrthoFinder遇到了类似的问题,见https://github.com/davidemms/OrthoF
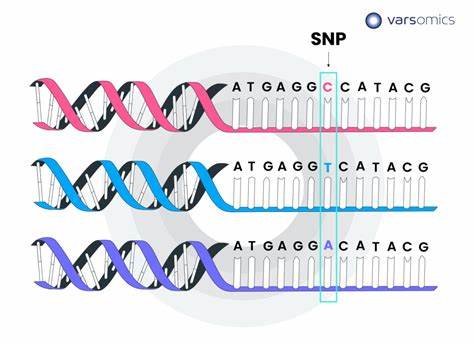
生信技能48 - 如何获取基因的SNP及RefSeq参考序列命名规则
1. SNP概念 SNP 是指基因组水平上由单个核苷酸的变异所引起的DNA 序列多态性,在群体中的发生频率不小于1 %,包括单个碱基的转换、颠换、插入和缺失等。每核苷酸发生突变的概率大约为10 -9 , 由于压力选择,SNP在单个基因和基因组以及动物不同种群间分布是不均匀的,在非编码区区SNP数量要多于编码区。 1.1 转换 转换是指同类型碱基之间的转换,如嘌呤与嘌呤( G2A) 、嘧啶与

生信软件21 - 多线程拆分NCBI-SRA文件工具pfastq-dump
在使用NCBI 工具fastq-dump拆分SRA文件时,拆分速度慢, fastq-dump拆分参数说明: –split-spot: 将双端测序分为两份,存放在同一个文件中–split-files: 将双端测序分为两份,存放在不同的文件,但是对于一方有而一方没有的reads直接丢弃–split-3 : 将双端测序分为两份,存放在不同的文件,但是对于一方有而一方没有的reads会单独放在一个文件
生信软件 | Sratools (操作SRA文件)
文章目录 1. 介绍2. 安装2.1 Conda 安装2.2 传统安装 3. 使用3.1 下载SRA3.2 抽取fastq文件 1. 介绍 Sratools是NCBI官方提供,用于操作SRA (reads and reference alignments) 数据的工具集合一般常用于下载SRA文件,从SRA文件中提取fastq,sam文件,查看SRA文件信息等 2. 安装 这
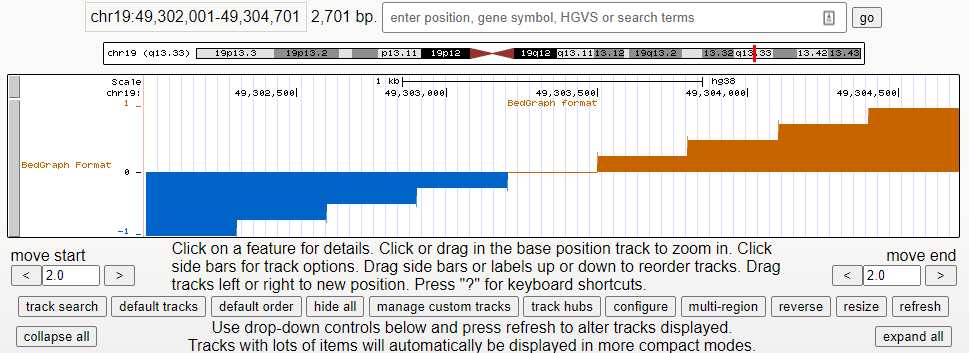
生信格式 | BedGraph(基因组浏览器绘制)
一、特点及适用场景: 存放区间的坐标轴信息和相关评分(score)的文件,主要用于存储稀疏,不连续的数据后缀名.bedGraph一般UCSC不建议采用该格式作为基因组浏览器输入文件,因为考虑到数据集大小与索引构建,都不如 bigwig 更高效,尤其在如果bedGraph数据集非常大(超过5000万行 ),推荐转为 bigwig 文件使用WigTobigWig将 bedGraph 转换为bigWi
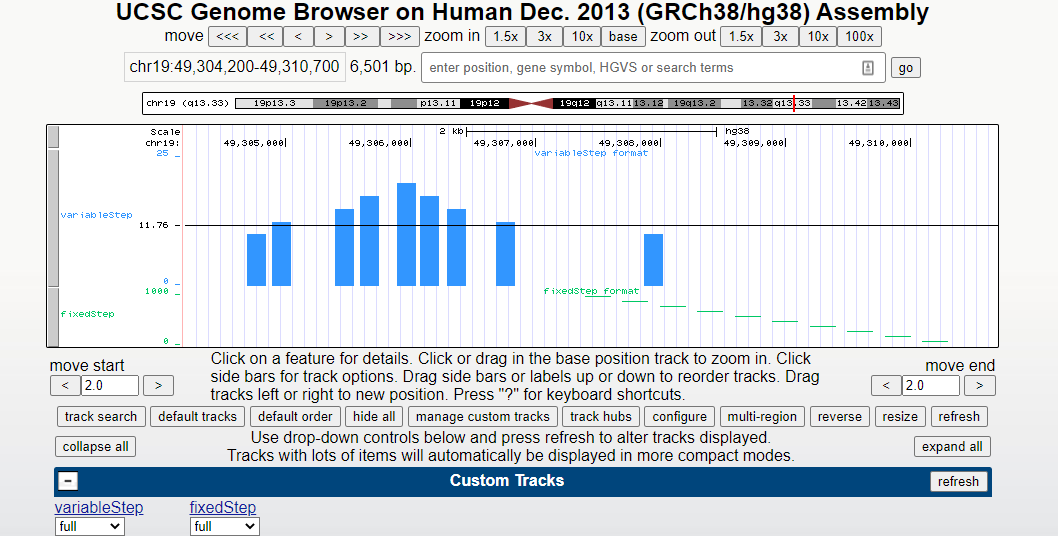
生信格式 | wig(基因组浏览器绘制)
文章目录 介绍一、variableStep 格式1、特点及适用场景:2、格式:3、例子: 二、fixedStep 格式1、特点及适用场景:2、格式:3、例子: 三、数据值例子 Wig,BigWig,BedGraph,这是几种在基因组浏览器上绘制图形的数据格式。 不同的数据格式可以满足不同的显示需求,下面我们一一来看: 介绍 wig 文件全称叫 Wiggle Tr