sra专题

WHU服务器下载SRA数据库方案
文章目录 一、你需要什么二、你要怎么做1. 准备下载索引2. wget -i list.txt 注意 提示:这里可以添加本文要记录的大概内容: 例如:随着人工智能的不断发展,机器学习这门技术也越来越重要,很多人都开启了学习机器学习,本文就介绍了机器学习的基础内容。 提示:以下是本篇文章正文内容,下面案例可供参考 一、你需要什么 177 地址下工作,一个明确的SRA

如何使用fastq-dump转换SRA格式
如何使用fastq-dump转换SRA格式 做生信的基本上都跟NCBI-SRA打过交道,尤其是fastq-dump大家肯定不陌生.NCBI的fastq-dump软件一直被大家归为目前网上文档做的最差的软件之一”,而我用默认参数到现在基本也没有出现过什么问题,感觉好像也没有啥问题, 直到今天看到如下内容, 并且用谷歌搜索的时候,才觉得大家对fastq-dump的评价非常很到位. 我们一般使用

如何修改已经公布的SRA数据的元信息
如何向NCBI的SRA上传数据介绍的是如何把本地的数据上传到SRA数据库上,这一篇介绍的是当你某天发现自己上传的数据的元信息中居然存在错误,比如说你把上传数据的单位写错了,那该如何修改呢? 写一份邮件给sra@ncbi.nlm.nih.gov,委托他们帮忙修改,一般2个工作日内都能搞定。 一定要注意,邮件的标题里面一定要有你上传数据的编号,例如SUB#,SRP#, SRS#, SRX#, SR

生信软件21 - 多线程拆分NCBI-SRA文件工具pfastq-dump
在使用NCBI 工具fastq-dump拆分SRA文件时,拆分速度慢, fastq-dump拆分参数说明: –split-spot: 将双端测序分为两份,存放在同一个文件中–split-files: 将双端测序分为两份,存放在不同的文件,但是对于一方有而一方没有的reads直接丢弃–split-3 : 将双端测序分为两份,存放在不同的文件,但是对于一方有而一方没有的reads会单独放在一个文件

生信软件 | Sratools (操作SRA文件)
文章目录 1. 介绍2. 安装2.1 Conda 安装2.2 传统安装 3. 使用3.1 下载SRA3.2 抽取fastq文件 1. 介绍 Sratools是NCBI官方提供,用于操作SRA (reads and reference alignments) 数据的工具集合一般常用于下载SRA文件,从SRA文件中提取fastq,sam文件,查看SRA文件信息等 2. 安装 这
sra数据下载linux,NCBI-SRA数据的下载方法
SRA 数据库: 为Sequence Read Archive 的缩写。主要存储高通量测序的原始数据,来自四个测序平台,分别为:Roche_LS454,Illumina,ABI_SOLID和HELICOS。从事生物信息分析的老师和同学一般都会接触SRA数据,下载SRA数据的方法也有很多,这里来简单总结一下。 一、SRA Tookit下载 选择需要的SRA Tookit 版本进行下载,下载后直接
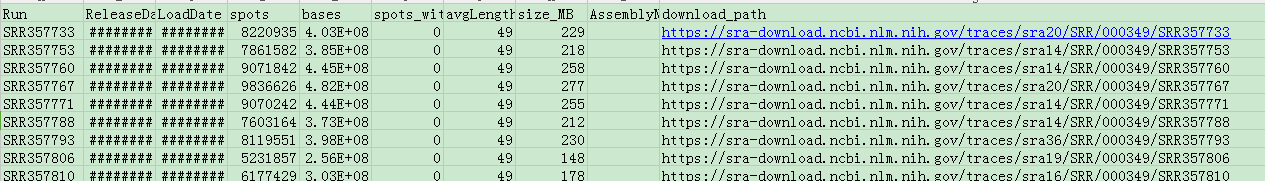
NCBI下载SRA数据
从NCBI下载数据本来是一件很简单的事情,但是今天碰到几个坑: 1、paper里没有提供SRA数据号、也没有提供路径; 2、不知道文件在ftp的地址,不能直接用wget下载 所以通过在NCBI官网,直接在SRA搜索栏里: 输入paper的title关键词NIFTY BGI 搜索结果: 选一个文件点击进去 进去之后,再点击SRP 然后: 出现如下内容: 然后选择所有SRR
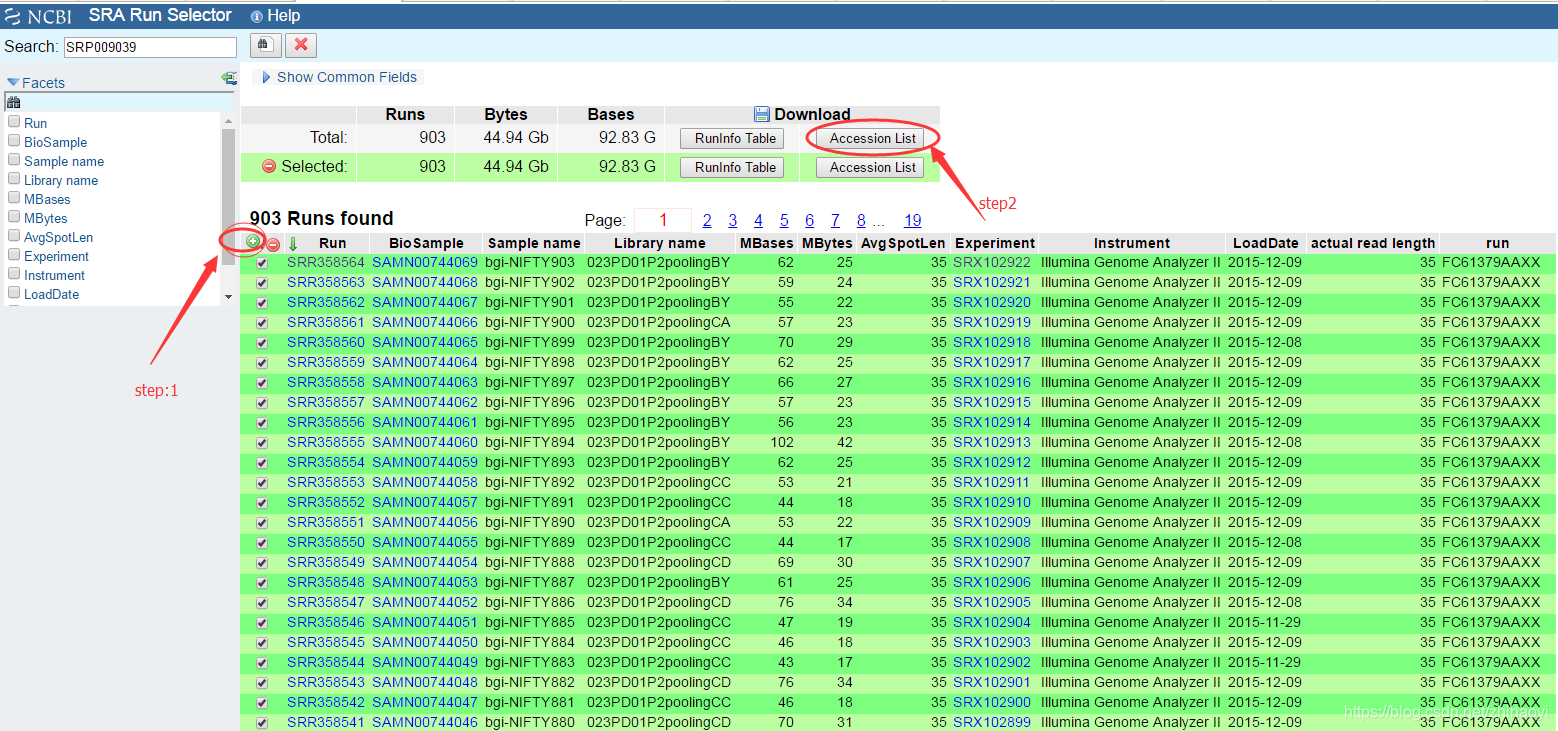
NCBI下载SRA数据(转)
从NCBI下载数据本来是一件很简单的事情,但是今天碰到几个坑: 1、paper里没有提供SRA数据号、也没有提供路径; 2、不知道文件在ftp的地址,不能直接用wget下载 所以通过在NCBI官网,直接在SRA搜索栏里: 输入paper的title关键词NIFTY BGI 搜索结果: 选一个文件点击进去 进去之后,再点击SRP 然后: 出现如下内容: 然后选择所有SRR文件:

使用aspera下载SRA数据速度高达 下载中国gsa数据? ascp
转载自:秘籍 | 惊了,使用aspera下载SRA数据速度高达 291Mb/s - 简书 一、安装Aspera Connect 安装Linux版的Aspera Connect # 上面链接是最新版,因此下载的时候去官网复制最新的链接地址下载,否则可能会报错wget https://d3gcli72yxqn2z.cloudfront.net/connect_latest/v4/bin

从NBCI上下载大量细菌的SRA测序文件及其对应的fasta文件
第一步 从genome数据库中下载对应信息 打开NCBI,进入genome数据库从Custom resources下进入Microbes,再进入Browse microbial genomes输入需要的物种,可以进一步筛选(filter),然后download 第二步 提取csv结果文件中的biosmple列的biosample号和replicon列的chromesome号 replicon
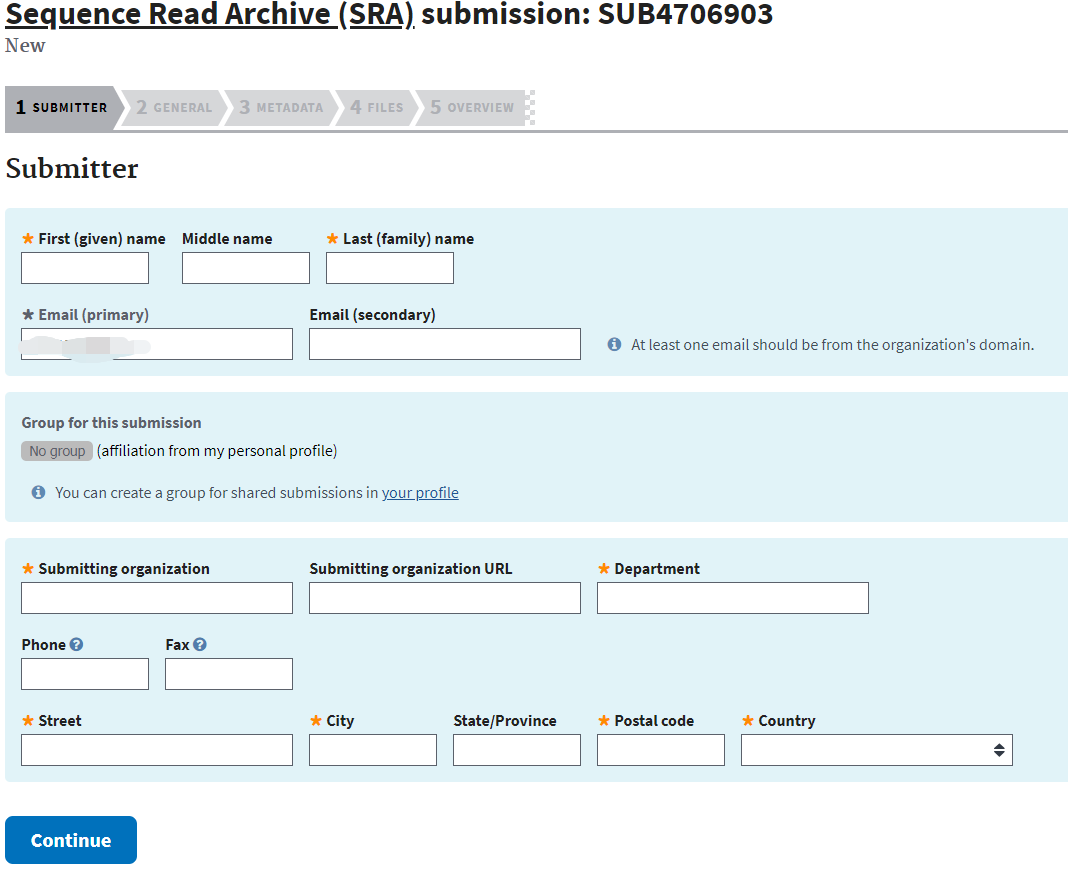
如何向NCBI的SRA上传数据
首先访问SRA的提交界面,https://submit.ncbi.nlm.nih.gov/subs/sra/,在没有登陆NCBI时,网页内容如下所示 未登录时 点击Log in, 会进入一个新的网页用于登陆或者创建新的NCBI账户。 登陆NCBI 在创建完账号或者登陆之后,返回之前的SRA上传页面,界面就变成了如下。 登陆后

SRA ToolKit (sra-tools) 的安装和使用
文章目录 前言从哪里下载 SRA ToolKit如何安装怎么用 前言 事情的起因是从NCBI SRA Database下载数据时的一个报错: path not found while resolving tree within virtual file system module - 'SRR17******' cannot be found 上次下载数据的时候还是上次,
从SRA数据下载开始学习ATACseq数据分析
1.打开NCBI GEO数据库找到你需要下载的数据页,打开SRA Run Selector,找到需要下载的原始数据的SRR编号。 2.创建一个txt文件并输入需要下载的SRR编号(好像SRA页面可以直接生成?待研究) vim sra.txt 3.prefetch命令进行数据下载(很慢,所以要放在后台下载,并且关闭shell窗口不影响) nohup prefetch
从SRA数据下载开始学习ATACseq数据分析
1.打开NCBI GEO数据库找到你需要下载的数据页,打开SRA Run Selector,找到需要下载的原始数据的SRR编号。 2.创建一个txt文件并输入需要下载的SRR编号(好像SRA页面可以直接生成?待研究) vim sra.txt 3.prefetch命令进行数据下载(很慢,所以要放在后台下载,并且关闭shell窗口不影响) nohup prefetch
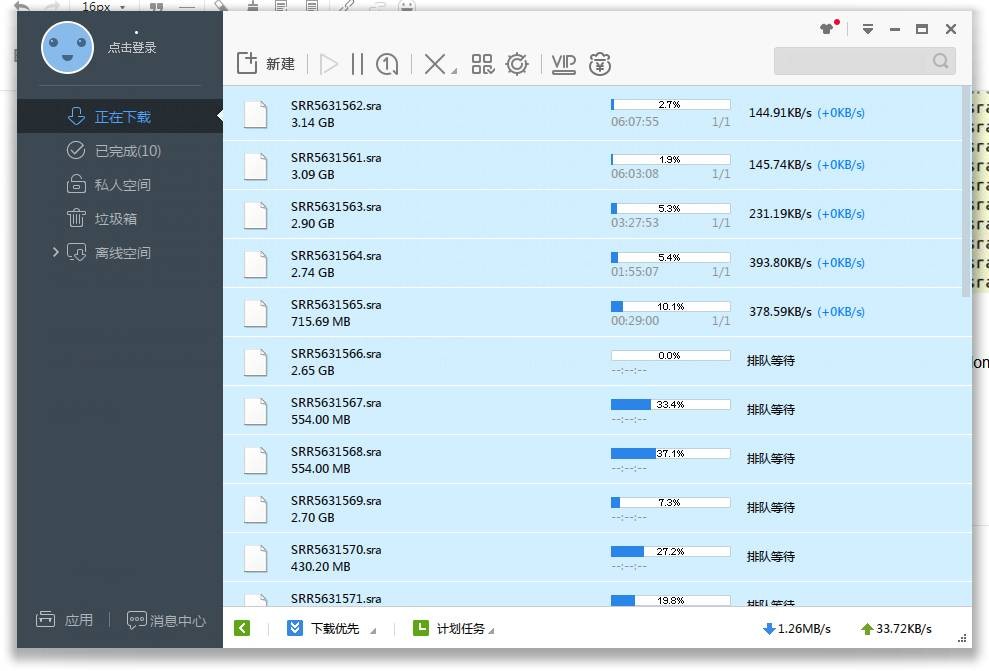
linux下载sra数据库,从NCBI当中SRA数据库中下载高通量测序数据
用别人的数据,发自己的文章 由于大多数杂志在文章发表前要求公开数据,所以随着测序文章的爆发,NCBI的SRA数据库当中积累了海量的测序数据。我们可以利用这些数据重新做数据挖掘,发表新的文章。 官方下载方法不太稳 要利用数据,首先得下载得到数据,虽然SRA数据库提供的SRA Toolkit 工具包里的prefetch可以下载,但是用这个方法下载数据需要经过复杂的设置,而且经常莫名奇妙的下不了,总的
linux下载sra数据库,从NCBI当中SRA数据库中下载高通量测序数据
用别人的数据,发自己的文章 由于大多数杂志在文章发表前要求公开数据,所以随着测序文章的爆发,NCBI的SRA数据库当中积累了海量的测序数据。我们可以利用这些数据重新做数据挖掘,发表新的文章。 官方下载方法不太稳 要利用数据,首先得下载得到数据,虽然SRA数据库提供的SRA Toolkit 工具包里的prefetch可以下载,但是用这个方法下载数据需要经过复杂的设置,而且经常莫名奇妙的下不了,总的
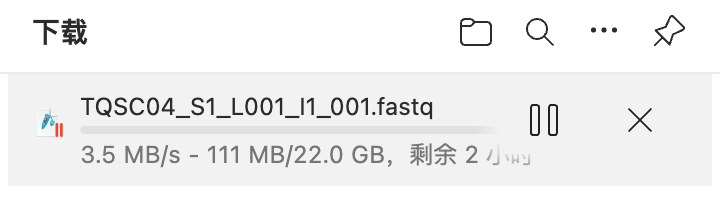
如何下载SRA存放在AWS的原始数据
通常,我们都是利用prefetch从NCBI上获取数据,然后用fasterp-dump/fastq-dump 转成fastq。但遗憾的SRA的数据是原数据的有损压缩,比如说我19年参与发表的文章里单细胞数据上传的是3个文件,但是当时的faster-dump/fastq-dump只能拆出2份(目前可以顺利拆出三份)。 但在https://trace.ncbi.nlm.nih.gov/Traces/
如何下载SRA存放在AWS的原始数据
通常,我们都是利用prefetch从NCBI上获取数据,然后用fasterp-dump/fastq-dump 转成fastq。但遗憾的SRA的数据是原数据的有损压缩,比如说我19年参与发表的文章里单细胞数据上传的是3个文件,但是当时的faster-dump/fastq-dump只能拆出2份(目前可以顺利拆出三份)。 但在https://trace.ncbi.nlm.nih.gov/Traces/
Linux系统中已知SRR号如何从NCBI上下载SRA数据到服务器中
高通量组学数据目前已经成为生物研究的重要板块,对于一些文章中出现的数据的挖掘尤其是人体数据的再利用也成为探究科学问题的重要前沿组成。通常情况下文章的高通量数据需要上传到NCBI的SRA(Sequence Read Archive)供大家下载学习,而我们也可以通过多种方法对数据进行下载再挖掘。在此介绍一种下载NCBI SRA数据的最佳方法。 首先,我们在下面的网址中
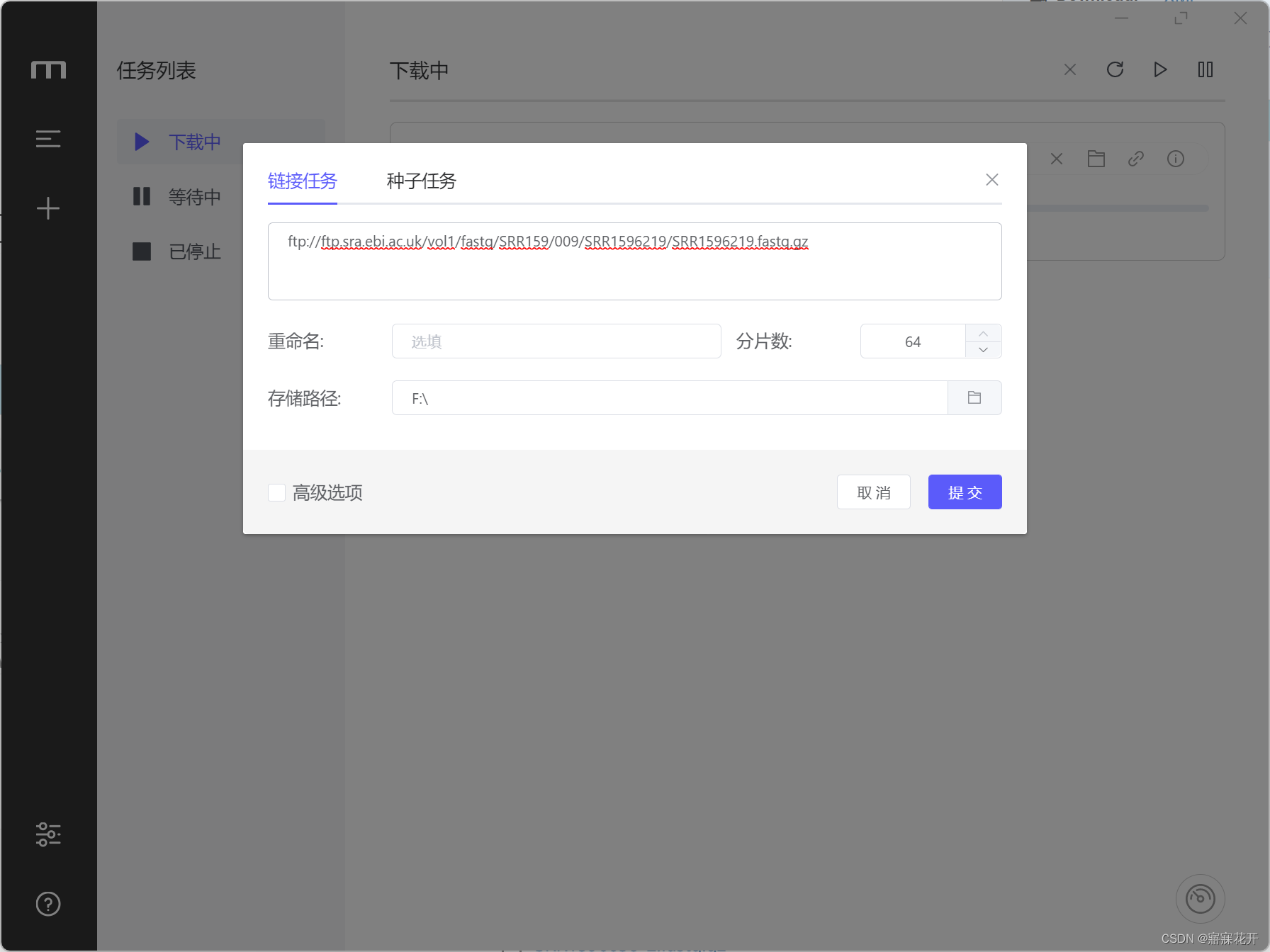
SRA原始数据下载-aspera-ascp命令Mortix
一、前置环境 win11子系统Ubuntu 20.0 安装aspera-cli (aspera的升级版) conda install -c hcc aspera-cli 安装后需要找到asperaweb_id_dsa.openssh /home/yang/miniconda3/pkgs/aspera-cli-3.9.6-h5e1937b_0/etc/asperaweb_id_dsa.o