本文主要是介绍从SRA数据下载开始学习ATACseq数据分析,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
1.打开NCBI GEO数据库找到你需要下载的数据页,打开SRA Run Selector,找到需要下载的原始数据的SRR编号。
2.创建一个txt文件并输入需要下载的SRR编号(好像SRA页面可以直接生成?待研究)
vim sra.txt3.prefetch命令进行数据下载(很慢,所以要放在后台下载,并且关闭shell窗口不影响)
nohup prefetch --option-file ./sra.txt &下载好之后会在本地出现以SRR编号命名的文件夹,需要进入其中的每一个文件进行解压
fasterq-dump -p -e 24 --split-3 *.sra可以看到每个.sra文件解压成了两个.fastq文件,分别是_1.fastq和 _2.fastq。
每个fastq文件内容格式有4行:第1行主要储存序列测序时的坐标等信息;第2行储存的是序列信息,正常情况都是用ATCG四个字母表示,但是当测序仪无法准确分辨该位置的序列信息时,会以N来代指此处的序列信息;第3行以“+”开始,可以存储一些信息,但是目前这一行都是空的;第4行存储的就是第2行每一个碱基的测序质量信息,其中的每一个符号所对应计算机的ASCII值是经过换算的phred值,而phred值等于33-10*logP,这里的P代表该位置测序发生错误的概率,简单来说,如果某个位置测得的序列十分可信,那么意味着该位置发生错误的概率极小,所以phred值就很大,即该值越大,说明测序的质量越好。
将所有生成的双端.fastq文件整合到一个文件夹中
4.fastqQC+multiQC
less /share/home/wuqian/job/atac_test/atac_test.txt |while read id;
do
fastqc ${id}_1.fastq -o /share/home/wuqian/job/atac_test/fastqc
fastqc ${id}_2.fastq -o /share/home/wuqian/job/atac_test/fastqc
done获得了.html的QC文件,此时可以吧这些文件拷贝到本地进行查看。
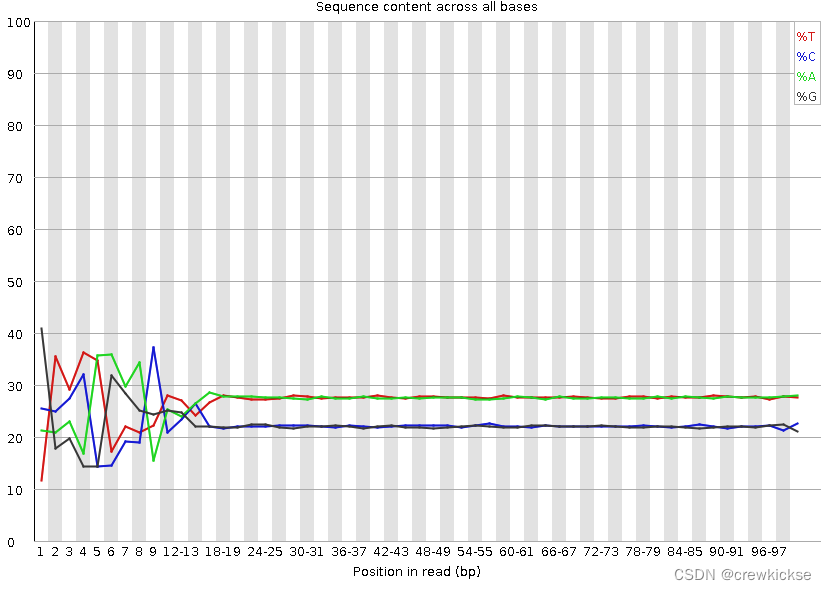
# 横轴是1 - 97 bp;纵轴是百分比
# 图中四条线代表A T C G在每个位置平均含量
# 理论上来说,A和T应该相等,G和C应该相等,但是一般测序的时候,刚开始测序仪状态不稳定,很可能出现上图的情况。像这种情况,即使测序的得分很高,也需要cut开始部分的序列信息,那根据这张图,我准备左边cut 18bp。根据质控报告对每个fastq文件进行质控
##conda 安装fastp
conda install -c bioconda fastp##创建一个质控文件夹
mkdir fastp##这时我们需要写一个循环来批量质控
##逻辑是,先读取我们之前的下载SRA的文件名,对每个名字的_1的fastq和_2的fastq利用fastp进行质控
## -f 从5‘剪切碱基数目; -t 从3‘剪切碱基数目
nohup
less /share/home/wuqian/job/atac_test/atac_test.txt |while read id;
do
fastp -i ${id}_1.fastq -o ./fastp/${id}_1.fastq -I ${id}_2.fastq -O ./fastp/${id}_2.fastq -f 18 -t 0 -L
done &##进入fastp文件夹,对处理过的数据利用fastqc重新进行质量分析
cd /share/home/wuqian/job/atac_test/fastp
mkdir fastqc_fastp##这时我们需要写一个循环来批量检查数据质量
##逻辑是,先读取我们之前的下载SRA的文件名,对每个名字的_1的fastq和_2的fastq进行检查
##-o的意思是fastqc输出的结果都输出到/home/xuyu/atac_ara/atac_data/fastp/fastqc_fastp文件夹内
nohup
less /share/home/wuqian/job/atac_test/atac_test.txt |while read id;
do
fastqc ${id}_1.fastq -o /share/home/wuqian/job/atac_test/fastp/fastqc_fastp
fastqc ${id}_2.fastq -o /share/home/wuqian/job/atac_test/fastp/fastqc_fastp
done &##将.html文件重新拷贝到本地查看QC结果5.reads mapping 到 genome
对测序获得的reads进行mapping,才能知道基因组上哪些区域富集比较多的reads,也就是开放的染色质区域。
mkdir genome
cd genome##下载基因组注释文件
nohup wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/001/635/GCF_000001635.27_GRCm39/GCF_000001635.27_GRCm39_genomic.fna.gz &gzip -d GCF_000001635.27_GRCm39/GCF_000001635.27_GRCm39_genomic.fna.gz##建立基因组索引
推荐直接在bowtie2官网下载对应的index文件,而不要尝试自己下载,下载完之后一定要知道索引文件放在什么位置#将去掉接头后的fastq文件比对会基因组,同时将sam文件转bam文件(因为sam文件太大,占用太多储存空间且不利于做大样本分析,因此需要转换为仅20-30%的bam文件),在bam文件进行sorted(按照基因组位置,这样有利于在IGV中查看变异)
##These parameters ensured that fragments up to 2 kb were allowed to align (-X2000) and that only unique aligning reads were collected (-m1).#!/bash/binbowtie2 -p 5 --very-sensitive -X 2000 -x /share/home/wuqian/job/atac_test/cleandata/index/mm9 -1 test_1.fastq -2 test_2.fastq > test.sam
提交bash脚本samtools view -bS test.sam| samtools sort -O bam -@ 5 -o - > test.raw.sorted.bam 6.对生成的bam文件进行数据清洗
##去除线粒体基因组序列,因为在线粒体基因组中没有感兴趣的ATAC-seq peaks
##只保留两条reads比对到同一条染色体(proper paired),还有高质量的比对结果(MAPQ>=30)nohup less /share/home/wuqian/job/atac_test/atac_test.txt |while read id;
do
samtools view -@ 5 -f 2 -q 30 -h ./sam_bam/${id}.bam | grep -v chrM | samtools sort -@ 4 -O bam -o ./sam_bam/${id}.rmChrM.bam
done >rmChrM.log 2>&1 &##去除PCR重复序列
##markdup -r意思是把重复的都删去 -t 4 意思是使用4线程处理 nohup less /share/home/wuqian/job/atac_test/atac_test.txt |while read id;
do
sambamba markdup -r -t 4 ${id}.rmChrM.bam ${id}_rmchrM_rmdup.bam
done >rmdup.log 2>&1 &##picard获取insert metric文件和条形图(shell脚本执行)
for i in `ls *.raw.sorted.bam`;
dosampleID="${i%.*}"echo "${sampleID}"java -Xmx32g -jar /share/home/wuqian/job/atac_test/picard.jar CollectInsertSizeMetrics I=$i OUTPUT=${sampleID}_insertMetrics H=${sampleID}_histo.pdf
doneecho $?##Tn5偏移同时将清洗之后的bam文件转为bed文件(shell脚本执行)
for i in `ls *.rmchrM.rmdup.bam`;
dosampleID="${i%.*}"echo "${sampleID}"bedtools bamtobed -i $i | awk 'BEGIN {OFS = "\t"} ; {if ($6 == "+") print $1, $2 + 5, $3 + 5, $4, $5, $6; else print $1, $2 - 4, $3 - 4, $4, $5, $6}' > ${sampleID}.bed
doneecho $?
echo 'You have bed files!'##将ENCODE黑名单中的基因去除
for i in `ls *.bed`;
dosampleID="${i%.*}"echo "${sampleID}"bedtools subtract -a $i -b mm9.blacklist.bed > ${sampleID}_debl.bed
doneecho $?
echo 'You have bed files without blacklist regions!'7.Peak Calling
##???对比对后的bam文件进行测序文库复杂度检验
##statc_qc.sh脚本
less /share/home/wuqian/job/atac_test/atac_test.txt | while read id;
do bedtools bamtobed -bedpe -i ${id}_rmchrM_rmdup.bam | \awk 'BEGIN{OFS="\t"}{print $1,$2,$4,$6,$9,$10}' | sort | uniq -c | \awk 'BEGIN{mt=0;m0=0;m1=0;m2=0} ($1==1){m1=m1+1} ($1==2){m2=m2+1} {m0=m0+1} {mt=mt+$1} END{m1_m2=-1.0; if(m2>0) m1_m2=m1/m2;printf "%d\t%d\t%d\t%d\t%f\t%f\t%f\n",mt,m0,m1,m2,m0/mt,m1/m0,m1_m2}' > ${id%%.*}.nodup.pbc.qc;
donenohup bash stat_qc.sh &##输出两个文件
SRR8528251.nodup.pbc.qc
SRR8528255.nodup.pbc.qccat一下查看NRF、PBC1、PBC2值,均符合
标准为:NRF>0.9, PBC1>0.9, and PBC2>10.##将bam文件转为bed文件
nohup less /share/home/wuqian/job/atac_test/atac_test.txt |while read id; do bedtools bamtobed -i ${id}_rmchrM_rmdup.bam > ${id%%.*}.raw.bed ;done >bed.log 2>&1 &
##对于chip_seq的数据分析,拿到bam文件之后直接peak calling就可以了,对于ATAC_seq而言,一定要偏移之后再进行peak calling
##在peak calling之前首先需要将Tn5转座酶插入位点进行偏移
##对于正链上的reads需要向右偏移4bp, 比对的起始位置加4,对于负链上的reads, 则向左偏移5bp, 比对的起始位置减5bp。##macs2 peak callingnohup ls *.last.bed | while read id; do (macs2 callpeak -t $id -f BED -n "${id%%.*}" -g mm --shift -100 --extsize 200 --nomodel) ;done &##macs2 callpeak会产生3个文件:NAME_peaks.xls, NAME_peaks.narrowPeak, NAME_summits.bed,其中最有用的文件是NAME_peaks.narrowPeak 是一个纯文本BED文件,列出了每个called peak的基因组坐标,以及各种统计数据(fold-change,p值和q值等)。8.下游可视化
(1)IGV可视化比对结果,需要bam文件转为bw文
##bed文件生成bam文件,sort后再生成bw文件
bedtools bedtobam -i test.bed -g mm9.chrom.sizes > test.last.bam
nohup samtools sort test.last.bam -@ 16 -O BAM -o test.final.bam >nohup.log 2>&1 &
samtools index test.final.bam##生成normalized(CPM)之后的bw文件
ls *.bam |while read id ; do
nohup bamCoverage --normalizeUsing CPM -b $id -o ${id%%.*}.bw &
done
(2)比较不同的peak文件
最常使用的工具是BEDTools,比如不同生物学重复之间可以发现共同的染色质区域:bedtools intersect ;实验组和对照组比较可以发现具有差异的染色质区域:bedtools subtract
(3)peak注释
CHIPseeker R包
(4)TF motif enrichment test
HOMER
It takes a peak file as input and checks for the enrichment of both known sequence motifs and de novo motifs
##UCSC官网下载对应的基因组注释文件,一定要是从一开始处理fastq文件开始就对应的注释文件,这里错误地使用mm39代替GRCm39,导致浪费了很多时间参数:findMotifsGenome.pl <peak/bed file> <genome file> <output directory>nohup findMotifsGenome.pl SRR8528251.bed /share/home/wuqian/job/atac_test/genome/GCF_000001635.27_GRCm39_genomic.fna motif_enrichment -preparse >motif.log 2>&1 & nohup findMotifsGenome.pl SRR8528255.bed /share/home/wuqian/job/atac_test/genome/GCF_000001635.27_GRCm39_genomic.fna motif_enrichment -preparse >motif.log 2>&1 &参考:https://github.com/harvardinformatics/ATAC-seq.git
明码标价之ATACseq|生信菜鸟团
「与国同庆,万字长文」ATAC-seq实战教程:从SRA数据下载到高分辨率论文主图绘制 - 徐寅生的文章 - 知乎 https://zhuanlan.zhihu.com/p/415718382
这篇关于从SRA数据下载开始学习ATACseq数据分析的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!





