seurat专题

Signac R|如何合并多个 Seurat 对象 (2)
引言 在本文中演示了如何合并包含单细胞染色质数据的多个 Seurat 对象。为了进行演示,将使用 10x Genomics 提供的四个 scATAC-seq PBMC 数据集: 500-cell PBMC 1k-cell PBMC 5k-cell PBMC 10k-cell PBMC 构建数据对象 接下来,将利用已经量化的矩阵数据,针对每个数据集构建一个 Seurat 数据对象
「Debug R」一个Seurat导致的Rstudio网页版经常被终止的bug
在网页版的Rstuio加载Seurat时,等待了很久都没有成功,刷新网页后就出现了如下的提示 报错信息 测试了其他包,例如ggplot2,都没有任何问题,唯独是Seurat出现了问题,因此我用关键词"seurat cause rsession terminated" 进行搜索,发现有人在Rstudio的社区上提出了这个问题,看来我并不是一个人遇到这个问题。 我尝试里帖子h
Seurat | 不同单细胞转录组的整合方法
一、涉及的新概念 参考(reference):将跨个体,跨技术,跨模式产生的不同的单细胞数据整合后的数据集 。也就是将不同来源的数据集组合到同一空间(reference)中。 从广义上讲,在概念上类似于基因组DNA序列的参考装配。 查询(query):单个实验产生的数据集 转化学习(transfer learning):产生一个于参考数据集(reference)上进行训练的模型,可以将信
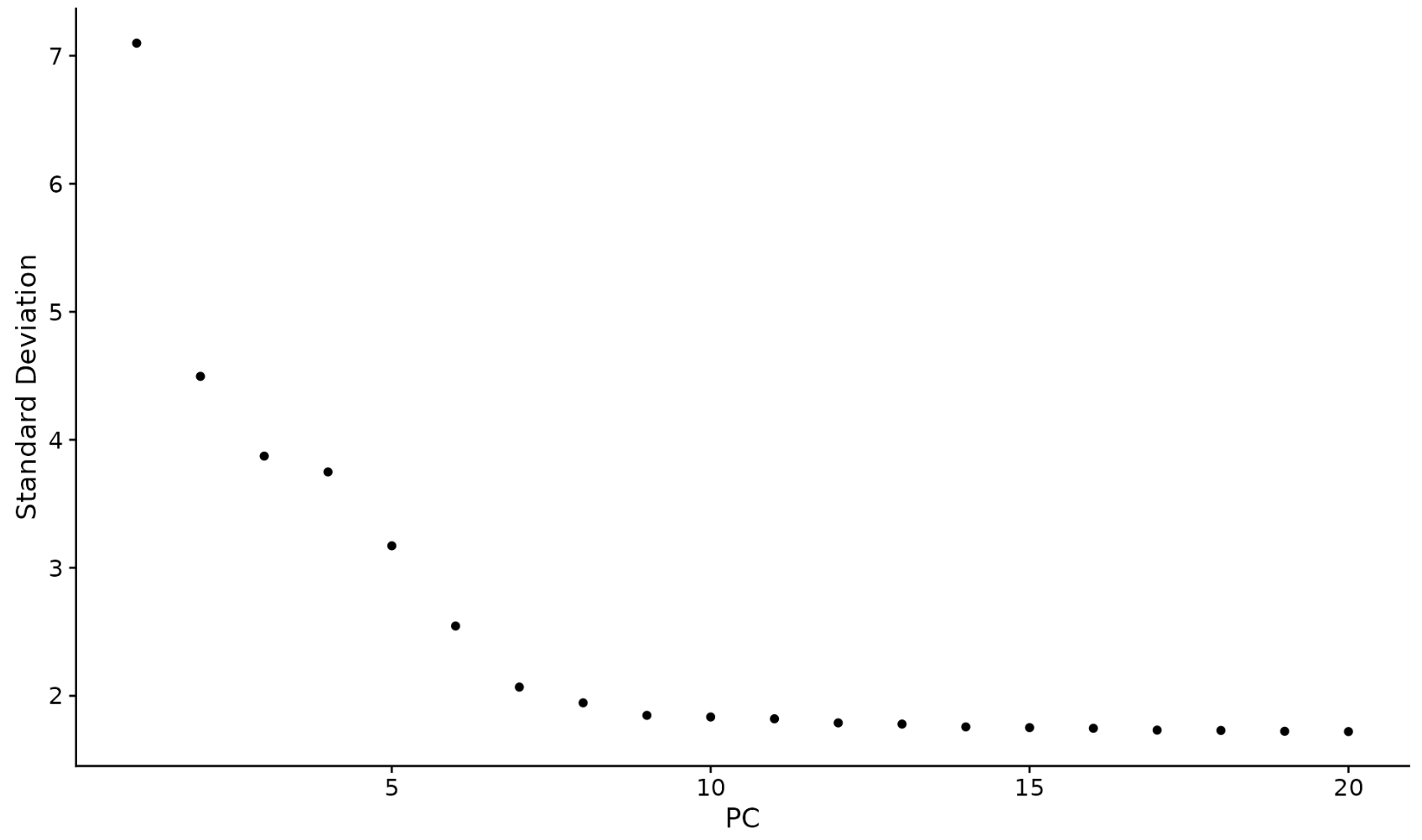
单细胞Seurat - 细胞聚类(3)
本系列持续更新Seurat单细胞分析教程,欢迎关注! 维度确定 为了克服 scRNA-seq 数据的任何单个特征中广泛的技术噪音,Seurat 根据 PCA 分数对细胞进行聚类,每个 PC 本质上代表一个“元特征”,它结合了相关特征集的信息。因此,顶部主成分代表了数据集的稳健压缩。但是,我们应该选择包含多少个成分? 10? 20? 100? 在 Macosko 等人中,我们实施了受 JackSt

R的seurat和python的scanpy对比学习
现在的单细胞分析,往往避免不了scanpy的使用,我们可以通过对比seurat来学习scanpy 今天的格式怎么都改不了。。。手机阅读有点费劲,,推荐电脑阅读。 单细胞数据分析概览 单细胞分析,总流程 python教程 seurat教程 seurat中与scanpy对等的函数操作 数据预处理 Seurat (R) Creat

Seurat包学习:如何查看R包函数源代码
我们很多时候都很好奇作者的r包是如何写出来的,手痒的时候就想学习一下源码,顺便改一改 问题来源 为什么要写今天这个推文呢? 起因是因为我想使用seurat自带函数画热图,奈何这个图不是那么好看 DoHeatmap(pbmc,features = features,draw.lines = FALSE ) 于是,我想自己手动改一下这个热图 p= DoHeatmap(pb
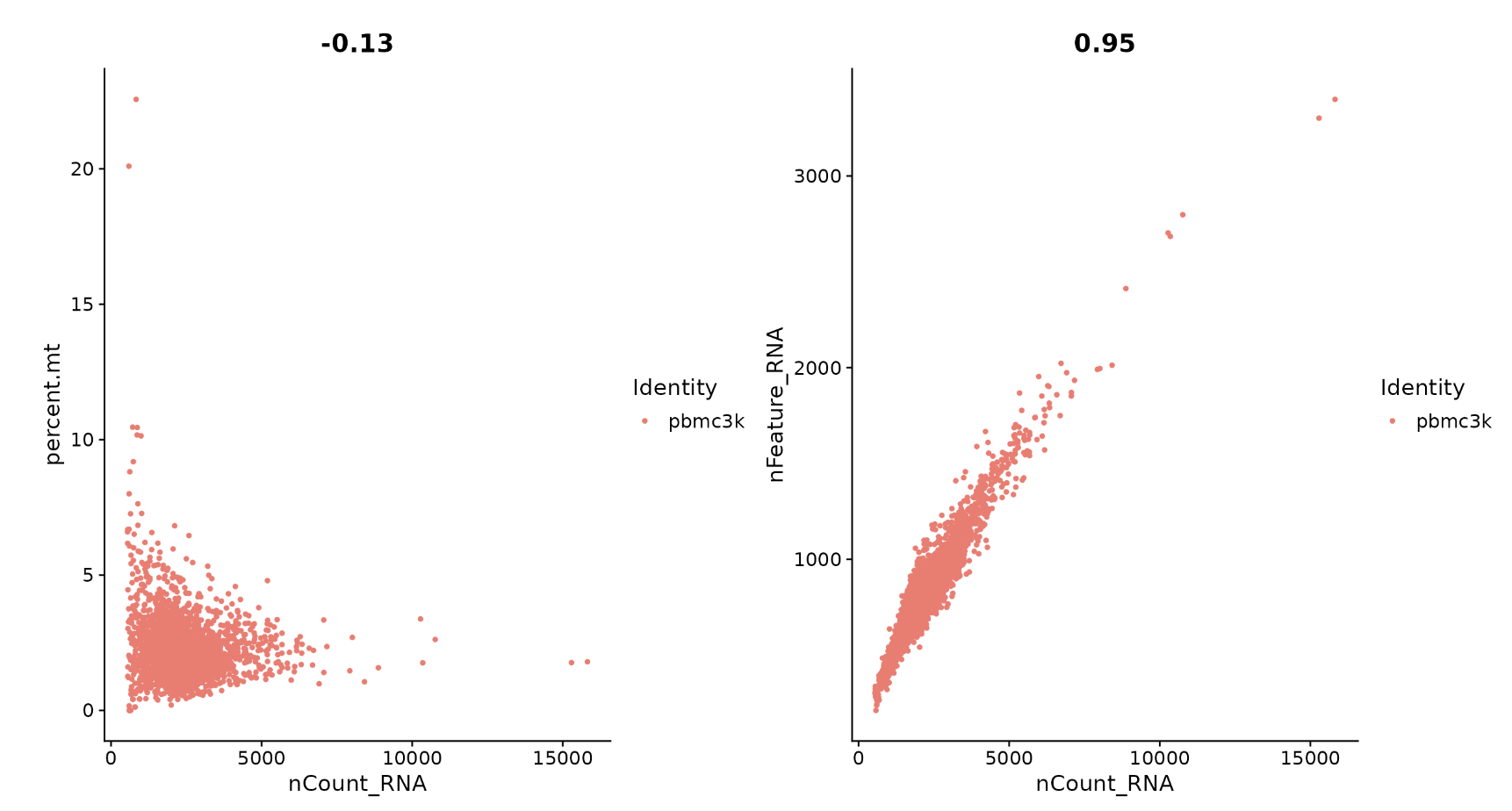
Seurat - 聚类教程 (1)
设置 Seurat 对象 在本教程[1]中,我们将分析 10X Genomics 免费提供的外周血单核细胞 (PBMC) 数据集。在 Illumina NextSeq 500 上对 2,700 个单细胞进行了测序。可以在此处[2]找到原始数据。 我们首先读取数据。 Read10X() 函数从 10X 读取 cellranger 管道的输出,返回唯一的分子识别 (UMI) 计数矩阵。该矩阵中的值表

【单细胞转录组】使用Seurat的Read10X函数读取10x文件时为什么有格式要求
非模式物种在分析时可能会遇到各种各样的问题,但是也能学习到一些不太会注意到的小细节。比如我在分析数据的时候,使用seurat查找差异基因后绘图,这里展示一下某个cluster的结果: 可以注意到出来Os号外还有一些看起来奇奇怪怪的基因,我本来以为是注释文件的问题。经师姐提醒,我去检查了一下features.tsv这个文件中的信息,发现这些长得奇奇怪怪的基因确实是存在的,而且是在第二列 也可
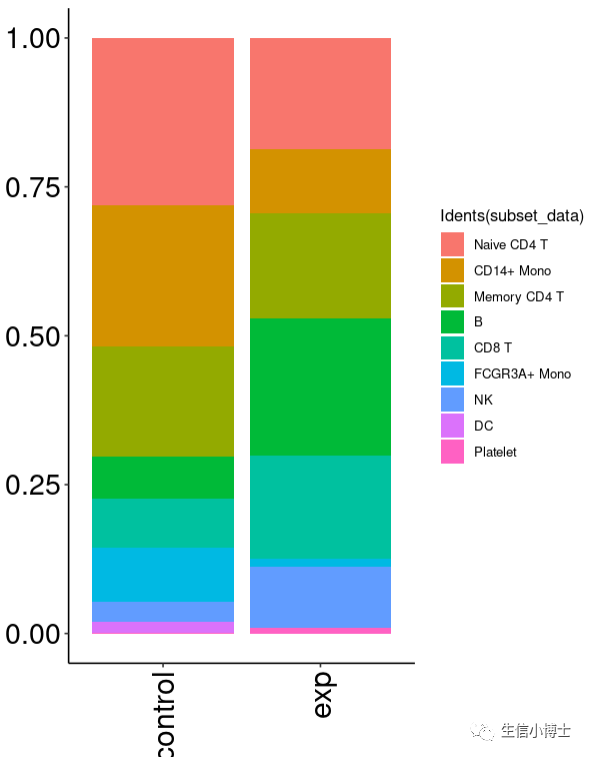
单细胞seurat-细胞比例分析-画图详细教程
大家好,今天我们来画单细胞中最简单的细胞比例图~ 1.老规矩,先加载pbmc数据 dir.create("~/gzh/细胞比例")setwd("~/gzh/细胞比例")subset_data=readRDS("~/gzh/pbmc3k_final.rds")table(stringr::str_split(string = colnames(subset_data),pattern = "
SCTransform normalization seurat
完成了前面的基础质控、过滤以及去除细胞周期的影响后,我们可以开始SCTransform normalization。 SCTransform normalization的优势: 1️⃣ 一个SCTransform函数即可替代NormalizeData, ScaleData, FindVariableFeatures三个函数;2️⃣ 对测序深度的校正效果要好于log标准化(10万以内的细胞都建
单细胞seurat入门—— 从原始数据到表达矩阵
根据所使用的建库方法,单细胞的RNA序列(也称为读取(reads)或标签(tags))将从转录本的3'端(或5'端)(10X Genomics,CEL-seq2,Drop-seq,inDrops)或全长转录本(Smart-seq)获得。 图片来源: Papalexi E and Satija R. Single-cell RNA sequencing to explore immun

R中seurat包函数FindMarkers报错Error in WhichCells.Seurat(object = object, idents = ident.1) :
在R中运行seurat包中的函数FindMarkers时,出现报错: Error in WhichCells.Seurat(object = object, idents = ident.1) : Cannot find the following identities in the object: 5 可能原因:seurat对象b已经被注释过,所以需要用注释后的名字,而不是最初以数

Seurat小提琴图为什么有的只有点儿如何给vlnplot加上肚子常看小提琴图vlnplot有多少个点细胞 加p值 加显著性zsf seurat对某个基因表达加显著性jitter抖动
library(Seurat) library(SeuratData) levels(pbmc3k.final) ** 通过添加参数map_signif_level=TRUE,可以将统计学差异表示为*符号。 https://cloud.tencent.com/developer/article/1692505 ** getwd()load("D:/Win10 System/Docume
SCS【4】单细胞转录组数据可视化分析 (Seurat 4.0)
点击关注,桓峰基因 桓峰基因的教程不但教您怎么使用,还会定期分析一些相关的文章,学会教程只是基础,但是如果把分析结果整合到文章里面才是目的,觉得我们这些教程还不错,并且您按照我们的教程分析出来不错的结果发了文章记得告知我们,并在文章中感谢一下我们哦! 公司英文名称:Kyoho Gene Technology (Beijing) Co.,Ltd. 桓峰基因公众号推出单细胞系列教程,有需要

seurat对象处理 找锚点
在找锚点合并之前,需要把每个seurat对象的细胞名改变成唯一 getwd() #改名字 #教程地址#https://cloud.tencent.com/developer/article/1697249#https://bioconductor.org/packages/release/data/experiment/vignettes/scRNAseq/inst/doc/scRNAseq.

Seurat -- Normalize Data
文章目录 brief为什么要做normalization 实例Normalization之前的预处理NormalizeData --> LogNormalize这种方法有是什么缺点呢 SCTransform仔细去看看这个函数所以这个函数到底干了什么这种normalization方法优秀在哪里正确使用scTransform brief seurat提供的教学里面包含了Stan
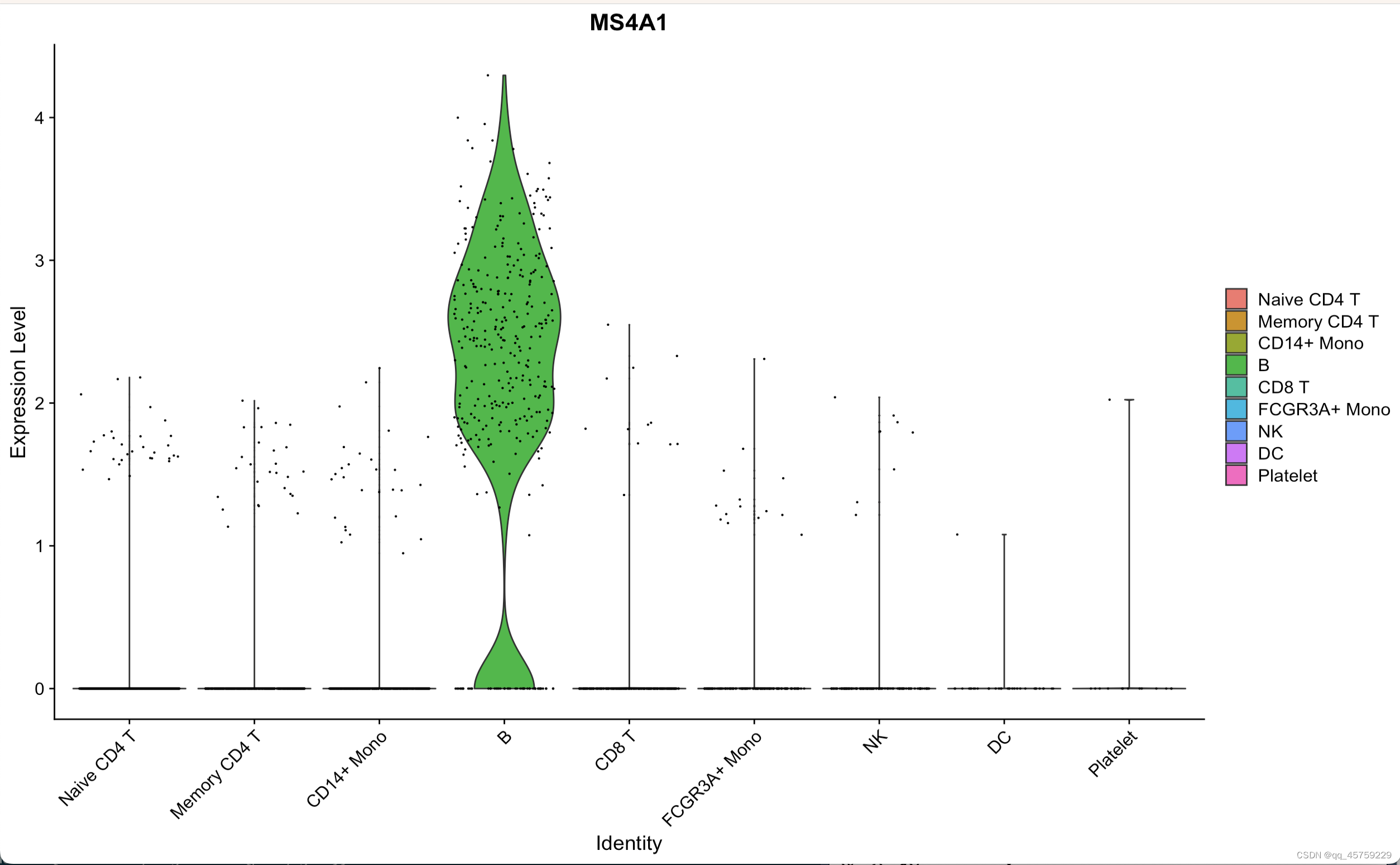
Seurat 源码学习之VlnPlot
今天很好奇Seurat里的Vlnplot是怎么画的,花了一个上午研究一下这个画图,其实还是很简单的哈, 以官网的pbmc3k为例 #library(dplyr)library(Seurat)library(patchwork)setwd("/Users/xiaokangyu/Desktop/Seurat_raw_code/")pbmc.data = Read10X("/Users/xi
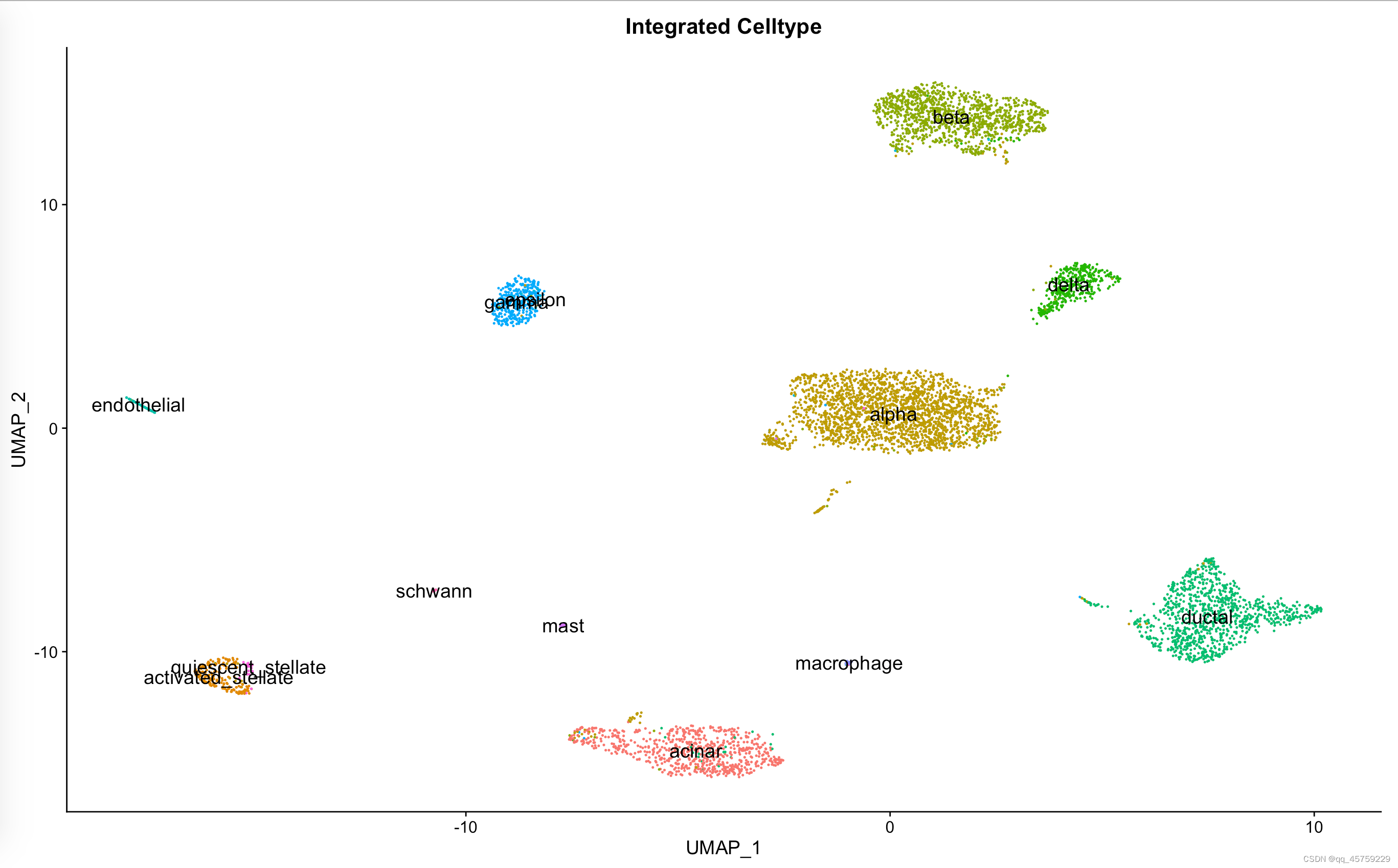
Seurat数据集处理流程
多数据集 pancreas数据集 suppressMessages(require(Seurat))suppressMessages(require(ggplot2))suppressMessages(require(cowplot))suppressMessages(require(scater))suppressMessages(require(scran))suppressMe
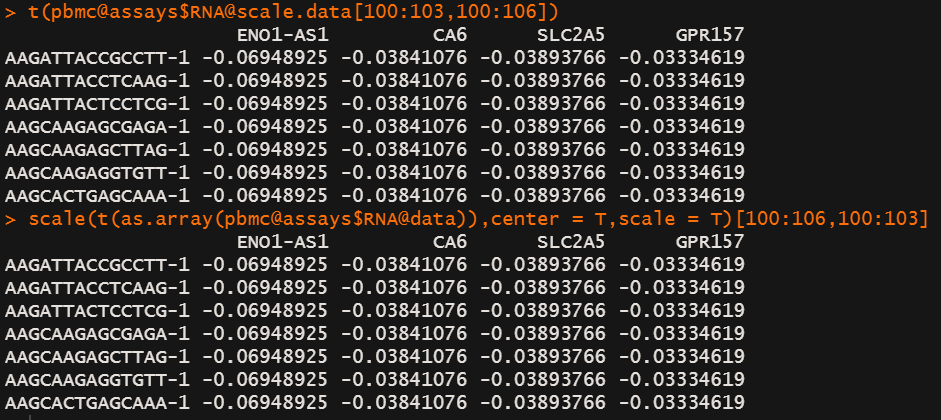
Seurat -- ScaleData学习
brief seurat提供了一个教学,其中global scale normalization之后又对数据进行了scale。 默认是对上一步 selected highly variable features进行scale。 概要图以及系列博文可以参见链接。 如果是 SCTransform则不需要手动运行这一步。 下面是就是教程提供的流程: library(dplyr)libra
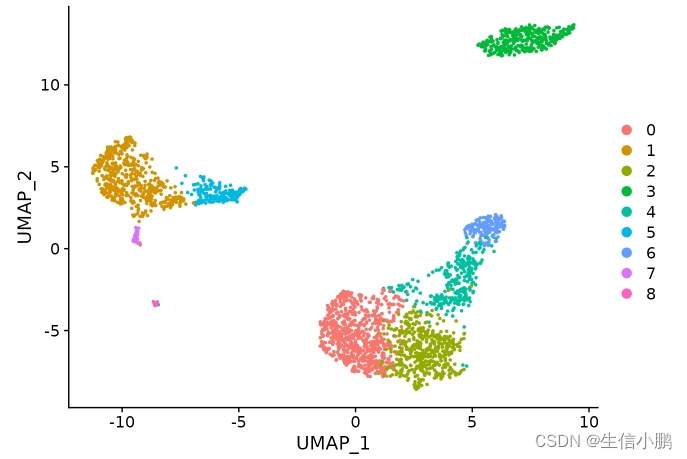
单细胞分析(一)——seurat包单个样本处理
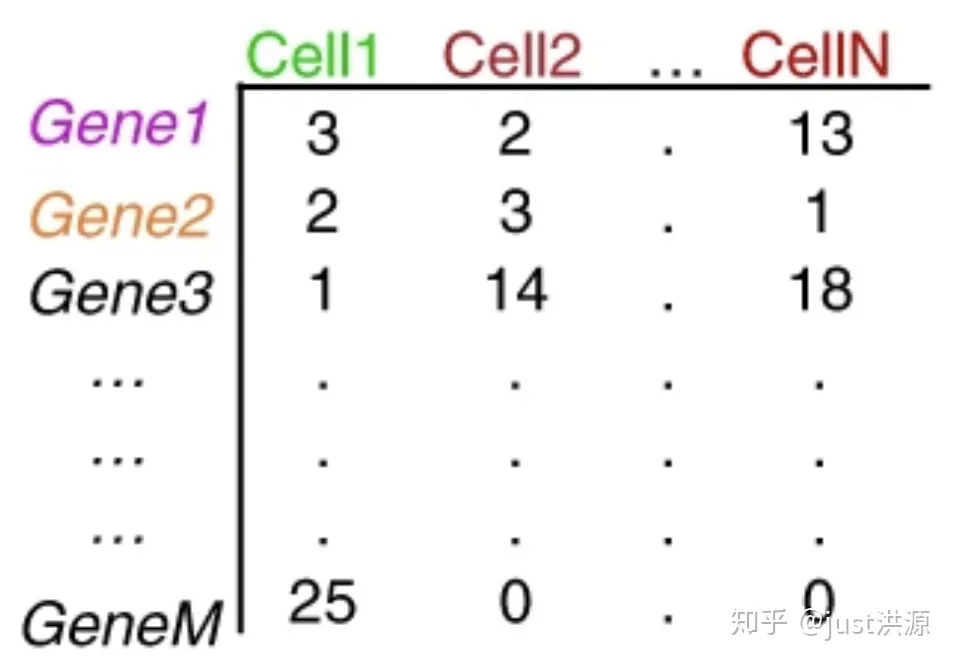
seurat包单个样本处理 10X genomics的基本原理创建对象加载数据创建 Seurat 对象count matrix是什么样子? 预处理流程计算线粒体含量质控展示正式筛选 数据标准化特征筛选寻找高变基因高变基因绘图 缩放数据PCA线性降维降维降维展示 UMAP非线性降维聚类分群参考文章 10X genomics的基本原理 大致如下 在这个教程中,主要将分析 10X Ge
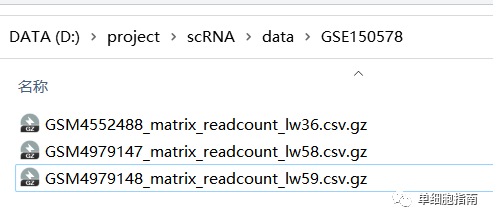
单细胞实战(1)数据下载-数据读取-seurat对象创建
这篇文章我们将介绍从geo数据库下载单细胞测序数据后,多种数据格式多样本情况下,如何读取数据并创建seurat对象。 本文主要结构: 一、数据下载 二、数据读取与seurat对象创建 单样本情况下各种格式数据的读取,读取后seurat对象的创建多样本情况下各种格式数据的读取,读取后seurat对象的创建、合并 一、数据下载 大家自行去GEO官网(https://www.ncbi.nlm