本文主要是介绍Seurat小提琴图为什么有的只有点儿如何给vlnplot加上肚子常看小提琴图vlnplot有多少个点细胞 加p值 加显著性zsf seurat对某个基因表达加显著性jitter抖动,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
library(Seurat)
library(SeuratData)
levels(pbmc3k.final)
**
通过添加参数map_signif_level=TRUE,可以将统计学差异表示为*符号。
https://cloud.tencent.com/developer/article/1692505
**
getwd()load("D:/Win10 System/Documents/WeChat Files/wxid_f27yna03e0v622/FileStorage/File/2022-11/merge.Rdata")library(Seurat)
head(merge@meta.data)
dim(merge) #[1] 15499 10979
table(merge$stim)
DimPlot(merge,label = T,group.by = "anno")
VlnPlot(merge,features = "FPR1")merge=FindVariableFeatures(merge,nfeatures = nrow(merge))
Assays(merge)
slotNames(merge)
head(merge@assays$RNA@data)
DotPlot(merge,features = "Fpr1")
which(rownames(merge)=="Fpr1")
rownames(merge)
Idents(merge)=merge$annop=VlnPlot(merge,features = "Fpr1",split.by = 'stim') #+geom_split_violin()
head(p)
mydata=p$data
head(mydata)
head(mydata)
library(ggplot2)
library(ggsignif)
library(ggpubr)
library(devtools)
install_github("JanCoUnchained/ggunchained")
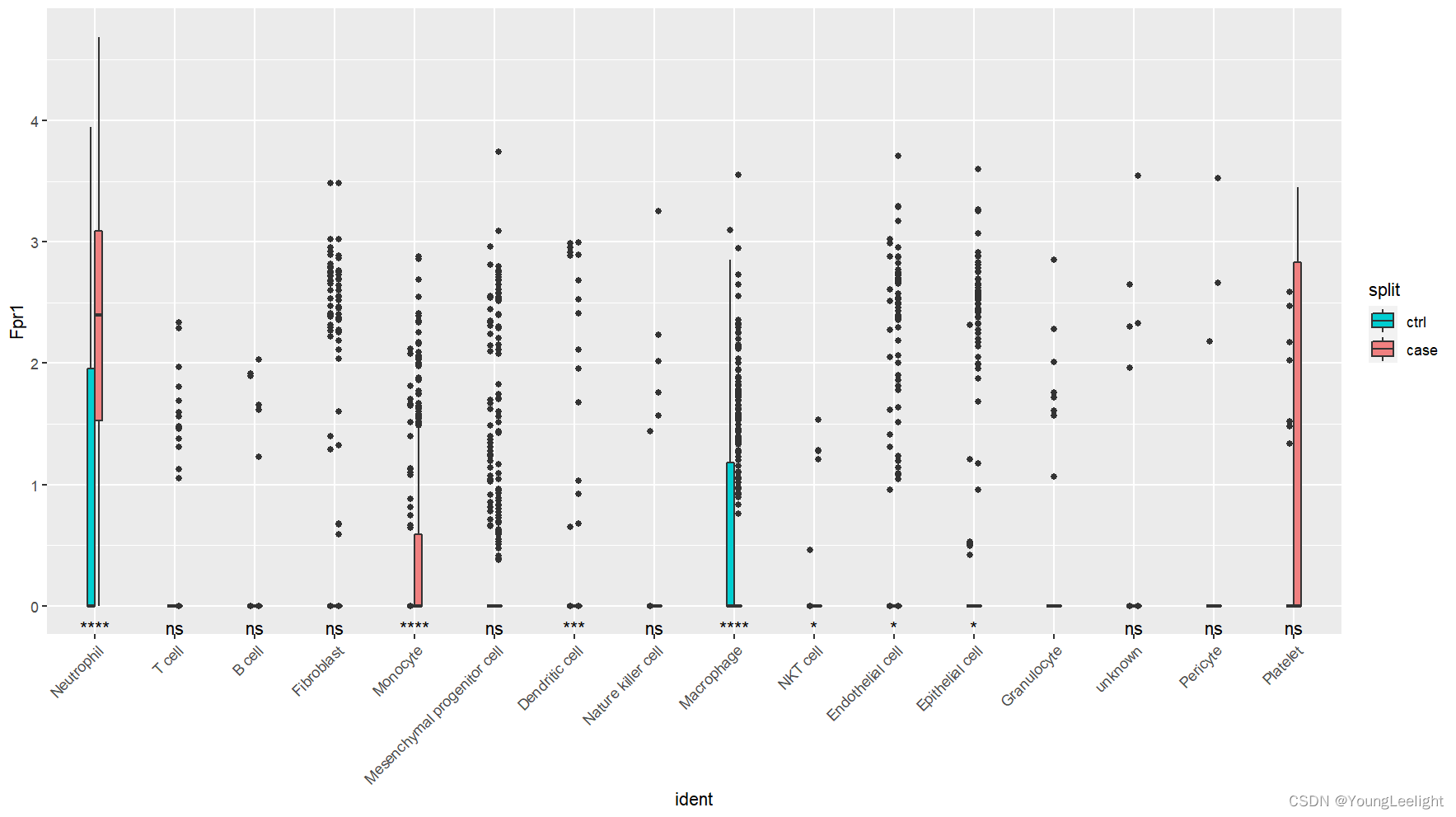
library(ggunchained) VlnPlot(subset(merge,Fpr1 > 0 ), "Fpr1",adjust = .2)+ #ggplot2::geom_violin()+stat_compare_means(mapping = p$data, aes(group = split),label="p.signif", label.y.npc = "bottom", vjust = 2)+theme_bw()ggplot(p$data,aes(ident,Fpr1, fill = split)) + geom_split_violin(scale = "width", adjust =.02, trim = TRUE) +geom_jitter()+ stat_compare_means(aes(group = split),label="p.signif", label.y.npc = "bottom", vjust = 2)+scale_fill_manual(values = c("#00CED1", "#F08080"))+RotatedAxis()+
theme_bw()ggplot(mydata, aes(x = ident, y = Fpr1, fill = split))+# geom_split_violin(scale = "width", alpha=0.7)+#ggplot2::geom_violin()+geom_jitter()+geom_boxplot(width = 0.2)+stat_compare_means(aes(group = split),label="p.signif", label.y.npc = "bottom", vjust = 2)+scale_fill_manual(values = c("#00CED1", "#F08080"))+RotatedAxis()# geom_signif(comparisons = compire,step_increase=0.1,map_signif_level = T,test = wilcox.test, textsize = 3)+theme(panel.grid.major=element_line(colour=NA),panel.background = element_rect(fill = "transparent",colour = "black"),plot.background = element_rect(fill = "transparent",colour = NA),panel.grid.minor = element_blank(), text = element_text(size = 12),axis.title = element_text(face="bold", size = 14),axis.text.x=element_text(size = 10, angle = 45, hjust = 1),axis.text=element_text(color = "black"), plot.title = element_text(size = 12, face="bold", hjust=0.5))+scale_y_continuous(name = "CSF1R expression")+scale_x_discrete(name = NULL)ggplot(data.use, aes(x = label, y = score, fill = type))+geom_split_violin(scale = "width", alpha=0.7)+geom_boxplot(width = 0.2)+stat_compare_means(aes(group = type),label="p.signif", label.y.npc = "bottom", vjust = 2)+scale_fill_manual(values = c("#00CED1", "#F08080"))+ geom_signif(comparisons = compire,step_increase=0.1,map_signif_level = T,test = wilcox.test, textsize = 3)+theme(panel.grid.major=element_line(colour=NA),panel.background = element_rect(fill = "transparent",colour = "black"),plot.background = element_rect(fill = "transparent",colour = NA),panel.grid.minor = element_blank(), text = element_text(size = 12),axis.title = element_text(face="bold", size = 16),axis.text.x=element_text(size = 10, angle = 45, hjust = 1),axis.text=element_text(color = "black"), plot.title = element_text(size = 18, face="bold", hjust=0.5))+ ggtitle(toupper("hallmark angiogenesis"))+scale_y_continuous(name = "SCORE", breaks=seq(-0.5,0.6,0.2), labels = seq(-0.5,0.6,0.2))+scale_x_discrete(name = NULL)geom_split_violin <- function(mapping = NULL,data = NULL,stat = "ydensity",position = "identity",...,draw_quantiles = NULL,trim = TRUE,scale = "area",na.rm = FALSE,show.legend = NA,inherit.aes = TRUE) {ggplot2::layer(data = data, mapping = mapping, stat = stat, geom = GeomSplitViolin,position = position, show.legend = show.legend, inherit.aes = inherit.aes,params = list(trim = trim, scale = scale, draw_quantiles = draw_quantiles,na.rm = na.rm, ...))
}#' @format NULL
#' @usage NULL
GeomSplitViolin <- ggplot2:::ggproto("GeomSplitViolin",ggplot2::GeomViolin,draw_group = function(self, data, ..., draw_quantiles = NULL) {data <- transform(data, xminv = x - violinwidth * (x - xmin), xmaxv = x + violinwidth * (xmax - x))grp <- data[1, "group"]newdata <- plyr::arrange(transform(data, x = if (grp %% 2 == 1) xminv else xmaxv), if (grp %% 2 == 1) y else -y)newdata <- rbind(newdata[1, ], newdata, newdata[nrow(newdata), ], newdata[1, ])newdata[c(1, nrow(newdata) - 1, nrow(newdata)), "x"] <- round(newdata[1, "x"])if (length(draw_quantiles) > 0 & !scales::zero_range(range(data$y))) {stopifnot(all(draw_quantiles >= 0), all(draw_quantiles <=1))quantiles <- ggplot2:::create_quantile_segment_frame(data, draw_quantiles)aesthetics <- data[rep(1, nrow(quantiles)), setdiff(names(data), c("x", "y")), drop = FALSE]aesthetics$alpha <- rep(1, nrow(quantiles))both <- cbind(quantiles, aesthetics)quantile_grob <- GeomPath$draw_panel(both, ...)ggplot2:::ggname("geom_split_violin", grid::grobTree(GeomPolygon$draw_panel(newdata, ...), quantile_grob))}else {ggplot2:::ggname("geom_split_violin", GeomPolygon$draw_panel(newdata, ...))}}
)[1] “Naive CD4 T” “Memory CD4 T” “CD14+ Mono” “B” “CD8 T”
[6] “FCGR3A+ Mono” “NK” “DC” “Platelet”
VlnPlot(pbmc3k.final, “CD4”,slot = “data”)

作为一个生物信息工程师,看到这样的图,请解释。
为什么CD14+ Mono和 Memory CD4 T 有怎么多的点,却没有小提琴呢?
那, 我们要看看作图细节了。
VlnPlot
function (object, features, cols = NULL, pt.size = 1, idents = NULL,
sort = FALSE, assay = NULL, group.by = NULL, split.by = NULL,
adjust = 1, y.max = NULL, same.y.lims = FALSE, log = FALSE,
ncol = NULL, combine = TRUE, slot = “data”, …)
{
return(ExIPlot(object = object, type = “violin”, features = features,
idents = idents, ncol = ncol, sort = sort, assay = assay,
y.max = y.max, same.y.lims = same.y.lims, adjust = adjust,
pt.size = pt.size, cols = cols, group.by = group.by,
split.by = split.by, log = log, slot = slot, combine = combine,
…))
}
<bytecode: 0x1a20fce0>
<environment: namespace:Seurat>`
可惜并没有细节,再看ExIPlot。
ExIPlot
Error: object ‘ExIPlot’ not found
不是显式函数啊。我们通过debug的方式进入函数内部。
debug(VlnPlot)
VlnPlot(pbmc3k.final, “CD4”,slot = “data”)
debugging in: VlnPlot(pbmc3k.final, “CD4”, slot = “data”)
debug: {
return(ExIPlot(object = object, type = “violin”, features = features,
idents = idents, ncol = ncol, sort = sort, assay = assay,
y.max = y.max, same.y.lims = same.y.lims, adjust = adjust,
pt.size = pt.size, cols = cols, group.by = group.by,
split.by = split.by, log = log, slot = slot, combine = combine,
…))
}
Browse[2]>
debug: return(ExIPlot(object = object, type = “violin”, features = features,
idents = idents, ncol = ncol, sort = sort, assay = assay,
y.max = y.max, same.y.lims = same.y.lims, adjust = adjust,
pt.size = pt.size, cols = cols, group.by = group.by, split.by = split.by,
log = log, slot = slot, combine = combine, …))
Browse[2]> ExIPlot
function (object, features, type = “violin”, idents = NULL, ncol = NULL,
sort = FALSE, assay = NULL, y.max = NULL, same.y.lims = FALSE,
adjust = 1, cols = NULL, pt.size = 0, group.by = NULL, split.by = NULL,
log = FALSE, combine = TRUE, slot = “data”, …)
{
assay <- assay %||% DefaultAssay(object = object)
DefaultAssay(object = object) <- assay
ncol <- ncol %||% ifelse(test = length(x = features) > 9,
yes = 4, no = min(length(x = features), 3))
data <- FetchData(object = object, vars = features, slot = slot)
features <- colnames(x = data)
if (is.null(x = idents)) {
cells <- colnames(x = object)
}
else {
cells <- names(x = Idents(object = object)[Idents(object = object) %in%
idents])
}
data <- data[cells, , drop = FALSE]
idents <- if (is.null(x = group.by)) {
Idents(object = object)[cells]
}
else {
object[[group.by, drop = TRUE]][cells]
}
if (!is.factor(x = idents)) {
idents <- factor(x = idents)
}
if (is.null(x = split.by)) {
split <- NULL
}
else {
split <- object[[split.by, drop = TRUE]][cells]
if (!is.factor(x = split)) {
split <- factor(x = split)
}
if (is.null(x = cols)) {
cols <- hue_pal()(length(x = levels(x = idents)))
cols <- Interleave(cols, InvertHex(hexadecimal = cols))
}
else if (length(x = cols) == 1 && cols == “interaction”) {
split <- interaction(idents, split)
cols <- hue_pal()(length(x = levels(x = idents)))
}
else {
cols <- Col2Hex(cols)
}
if (length(x = cols) < length(x = levels(x = split))) {
cols <- Interleave(cols, InvertHex(hexadecimal = cols))
}
cols <- rep_len(x = cols, length.out = length(x = levels(x = split)))
names(x = cols) <- sort(x = levels(x = split))
}
if (same.y.lims && is.null(x = y.max)) {
y.max <- max(data)
}
plots <- lapply(X = features, FUN = function(x) {
return(SingleExIPlot(type = type, data = data[, x, drop = FALSE],
idents = idents, split = split, sort = sort, y.max = y.max,
adjust = adjust, cols = cols, pt.size = pt.size,
log = log, …))
})
label.fxn <- switch(EXPR = type, violin = ylab, ridge = xlab,
stop("Unknown ExIPlot type ", type, call. = FALSE))
for (i in 1:length(x = plots)) {
key <- paste0(unlist(x = strsplit(x = features[i], split = “"))[1],
"”)
obj <- names(x = which(x = Key(object = object) == key))
if (length(x = obj) == 1) {
if (inherits(x = object[[obj]], what = “DimReduc”)) {
plots[[i]] <- plots[[i]] + label.fxn(label = “Embeddings Value”)
}
else if (inherits(x = object[[obj]], what = “Assay”)) {
next
}
else {
warning("Unknown object type ", class(x = object),
immediate. = TRUE, call. = FALSE)
plots[[i]] <- plots[[i]] + label.fxn(label = NULL)
}
}
else if (!features[i] %in% rownames(x = object)) {
plots[[i]] <- plots[[i]] + label.fxn(label = NULL)
}
}
if (combine) {
combine.args <- list(plots = plots, ncol = ncol)
combine.args <- c(combine.args, list(…))
if (!“legend” %in% names(x = combine.args)) {
combine.args$legend <- “none”
}
plots <- do.call(what = “CombinePlots”, args = combine.args)
}
return(plots)
}
<bytecode: 0x19dc8580>
<environment: namespace:Seurat>
这一层函数也没讲小提琴如何画的,再看SingleExIPlot。
Browse[2]> SingleExIPlot
function (data, idents, split = NULL, type = “violin”, sort = FALSE,
y.max = NULL, adjust = 1, pt.size = 0, cols = NULL, seed.use = 42,
log = FALSE)
{
if (!is.null(x = seed.use)) {
set.seed(seed = seed.use)
}
if (!is.data.frame(x = data) || ncol(x = data) != 1) {
stop(“'SingleExIPlot requires a data frame with 1 column”)
}
feature <- colnames(x = data)
dataKaTeX parse error: Expected 'EOF', got '&' at position 49: …cter(x = sort) &̲& nchar(x = sor…ident <- factor(x = data i d e n t , l e v e l s = n a m e s ( x = r e v ( x = s o r t ( x = t a p p l y ( X = d a t a [ , f e a t u r e ] , I N D E X = d a t a ident, levels = names(x = rev(x = sort(x = tapply(X = data[, feature], INDEX = data ident,levels=names(x=rev(x=sort(x=tapply(X=data[,feature],INDEX=dataident, FUN = mean), decreasing = grepl(pattern = paste0(“^”,
tolower(x = sort)), x = “decreasing”)))))
}
if (log) {
noise <- rnorm(n = length(x = data[, feature]))/200
data[, feature] <- data[, feature] + 1
}
else {
noise <- rnorm(n = length(x = data[, feature]))/1e+05
}
if (all(data[, feature] == data[, feature][1])) {
warning(paste0("All cells have the same value of ", feature,
“.”))
}
else {
data[, feature] <- data[, feature] + noise
}
axis.label <- “Expression Level”
y.max <- y.max %||% max(data[, feature])
if (is.null(x = split) || type != “violin”) {
vln.geom <- geom_violin
fill <- “ident”
}
else {
dataKaTeX parse error: Expected 'EOF', got '}' at position 82: …<- "split" }̲ switch(EXP…ident))
splits <- unique(x = as.vector(x = dataKaTeX parse error: Expected '}', got 'EOF' at end of input: …vector(x = datasplit))))
}
if (is.null(x = names(x = labels))) {
names(x = labels) <- labels
}
}
else {
labels <- levels(x = droplevels(data$ident))
}
plot <- plot + scale_fill_manual(values = cols, labels = labels)
}
return(plot)
}
<bytecode: 0x1a735330>
<environment: namespace:Seurat>
我们看到核心了,ggplot的代码。
geom <- list(vln.geom(scale = "width", adjust = adjust, trim = TRUE), theme(axis.text.x = element_text(angle = 45, hjust = 1)))jitter <- geom_jitter(height = 0, size = pt.size)log.scale <- scale_y_log10()axis.scale <- ylim
这句子写的真美啊。
那我们就要来试一下了。记得退出debug模式哦。
undebug(VlnPlot)
p<-VlnPlot(pbmc3k.final, “CD4”,slot = “data”)
#ggplot对象中记录了一张图的所有信息,为了方便演示,我们只取数据出来。
head(p$data) # 从图中抠数据,学会了吗?
CD4 ident
AAACATACAACCAC 1.370958e-05 Memory CD4 T
AAACATTGAGCTAC -5.646982e-06 B
AAACATTGATCAGC 3.631284e-06 Memory CD4 T
AAACCGTGCTTCCG 6.328626e-06 CD14+ Mono
AAACCGTGTATGCG 4.042683e-06 NK
AAACGCACTGGTAC -1.061245e-06 Memory CD4 T
原汁原味的啊:
ggplot(p$data,aes(ident,CD4)) + geom_violin() + theme_bw()
啥也没有:
加个抖动吧:
ggplot(p$data,aes(ident,CD4)) + geom_violin() + geom_jitter()+ theme_bw()
这下有点了。所以我们看到的点有左右的区分其实是抖出来的,本身数据的点应该是在一条直线上。然而,小提琴呢?
ggplot(p$data,aes(ident,CD4)) + geom_violin(scale = "width", adjust =1, trim = TRUE) + geom_jitter()+ theme_bw()
这里我们用seurat内部绘制小提琴图的方式还原了我们问题:为什么CD14+ Mono和 Memory CD4 T 有怎么多的点,却没有小提琴呢?经过上面演示我们知道,其实默认的情况下,我们的数据是都没有小提琴的。所以,当务之急是抓紧时间看看geom_violin的帮助文档。
?geom_violin
?geom_violin
?geom_violin
好了,我们知道一个关键的参数scale = "width"导致了这种局面,其他没有出现小提琴的应该是零值比例太多。
作为好奇,我们看看改一下adjust会有什么改变。
ggplot(p$data,aes(ident,CD4)) + geom_violin(scale = "width", adjust =.5, trim = TRUE) + geom_jitter()+ theme_bw()
腰变细了,好玩。
既然已经基本锁定问题,我们如何画出都有小提琴的小提琴图呢?也许可以用的方法之一就是,数据过滤。
VlnPlot(subset(pbmc3k.final,CD4 > 0 ), "CD4")+ theme_bw()
什么?改一下腰围?
VlnPlot(subset(pbmc3k.final,CD4 > 0 ), "CD4",adjust = .2)+ theme_bw()
Seurat小提琴图为什么有的只有点儿?那是因为还有更多的点没忽视。
#https://cloud.tencent.com/developer/article/1677923
这篇关于Seurat小提琴图为什么有的只有点儿如何给vlnplot加上肚子常看小提琴图vlnplot有多少个点细胞 加p值 加显著性zsf seurat对某个基因表达加显著性jitter抖动的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!