本文主要是介绍Seurat - 聚类教程 (1),希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
设置 Seurat 对象
在本教程[1]中,我们将分析 10X Genomics 免费提供的外周血单核细胞 (PBMC) 数据集。在 Illumina NextSeq 500 上对 2,700 个单细胞进行了测序。可以在此处[2]找到原始数据。
我们首先读取数据。 Read10X() 函数从 10X 读取 cellranger 管道的输出,返回唯一的分子识别 (UMI) 计数矩阵。该矩阵中的值表示在每个细胞(列)中检测到的每个特征(即基因;行)的分子数量。请注意,较新版本的 cellranger 现在也使用 h5 文件格式进行输出,可以使用 Seurat 中的 Read10X_h5() 函数读取该格式。
接下来我们使用计数矩阵来创建 Seurat 对象。该对象充当容器,其中包含单细胞数据集的数据(如计数矩阵)和分析(如 PCA 或聚类结果)。例如,在 Seurat v5 中,计数矩阵存储在 pbmc[["RNA"]]$counts 中。
library(dplyr)
library(Seurat)
library(patchwork)
# Load the PBMC dataset
pbmc.data <- Read10X(data.dir = "/brahms/mollag/practice/filtered_gene_bc_matrices/hg19/")
# Initialize the Seurat object with the raw (non-normalized data).
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
pbmc
-
输出
## An object of class Seurat
## 13714 features across 2700 samples within 1 assay
## Active assay: RNA (13714 features, 0 variable features)
## 1 layer present: counts
-
示例
# Lets examine a few genes in the first thirty cells
pbmc.data[c("CD3D", "TCL1A", "MS4A1"), 1:30]
# 输出
## 3 x 30 sparse Matrix of class "dgCMatrix"
##
## CD3D 4 . 10 . . 1 2 3 1 . . 2 7 1 . . 1 3 . 2 3 . . . . . 3 4 1 5
## TCL1A . . . . . . . . 1 . . . . . . . . . . . . 1 . . . . . . . .
## MS4A1 . 6 . . . . . . 1 1 1 . . . . . . . . . 36 1 2 . . 2 . . . .
矩阵中.的值代表 0(未检测到分子)。由于 scRNA-seq 矩阵中的大多数值都是 0,因此 Seurat 只要有可能就使用稀疏矩阵表示。这会显著节省 Drop-seq/inDrop/10x 数据的内存和速度。
dense.size <- object.size(as.matrix(pbmc.data))
dense.size
## 709591472 bytes
sparse.size <- object.size(pbmc.data)
sparse.size
## 29905192 bytes
dense.size/sparse.size
## 23.7 bytes
预处理
以下步骤涵盖 Seurat 中 scRNA-seq 数据的标准预处理工作流程。这些基于 QC 指标、数据标准化和缩放以及高度可变特征的检测的细胞选择和过滤。
Seurat 允许您轻松探索 QC 指标并根据任何用户定义的标准过滤细胞。常用的一些 QC 指标包括:
-
每个细胞中检测到的唯一(unique)基因的数量 -
低质量的细胞或空液滴通常含有很少的基因 -
细胞双联体或多联体可能表现出异常高的基因计数
-
-
同样,细胞内检测到的分子总数(与唯一(unique)基因密切相关) -
映射到线粒体基因组的读数百分比 -
低质量/垂死细胞通常表现出广泛的线粒体污染 -
我们使用 PercentageFeatureSet() 函数计算线粒体 QC 指标,该函数计算源自一组特征的计数百分比 -
我们使用以 MT- 开头的所有基因的集合作为线粒体基因的集合
-
# The [[ operator can add columns to object metadata. This is a great place to stash QC stats
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
-
Seurat 中的 QC 指标存储在哪里?
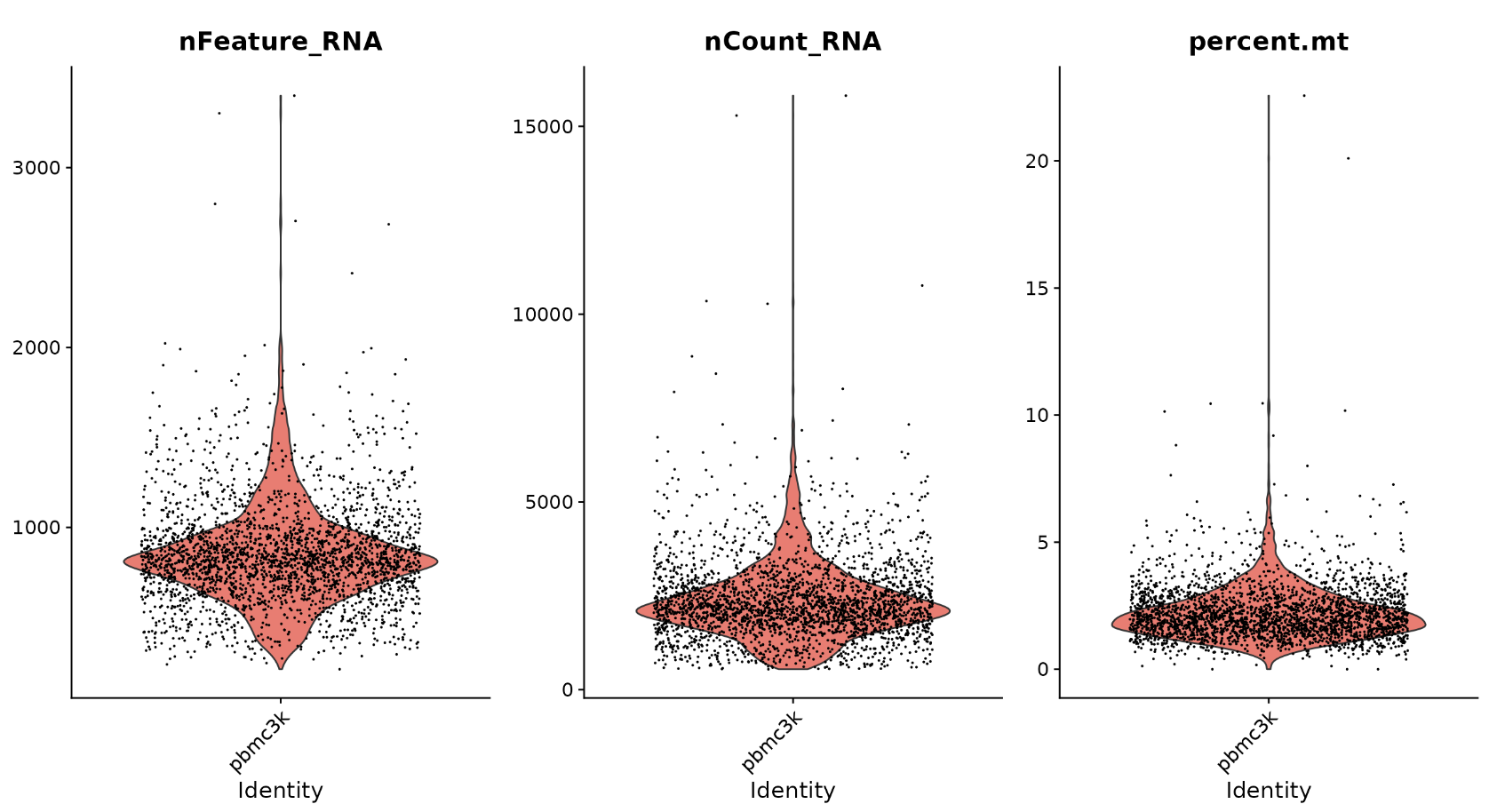
在下面的示例中,我们将 QC 指标可视化,并使用它们来过滤细胞。
我们过滤具有唯一特征计数超过 2,500 或少于 200 的细胞;我们过滤线粒体计数 >5% 的细胞
# Visualize QC metrics as a violin plot
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
# FeatureScatter is typically used to visualize feature-feature relationships, but can be used
# for anything calculated by the object, i.e. columns in object metadata, PC scores etc.
plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2
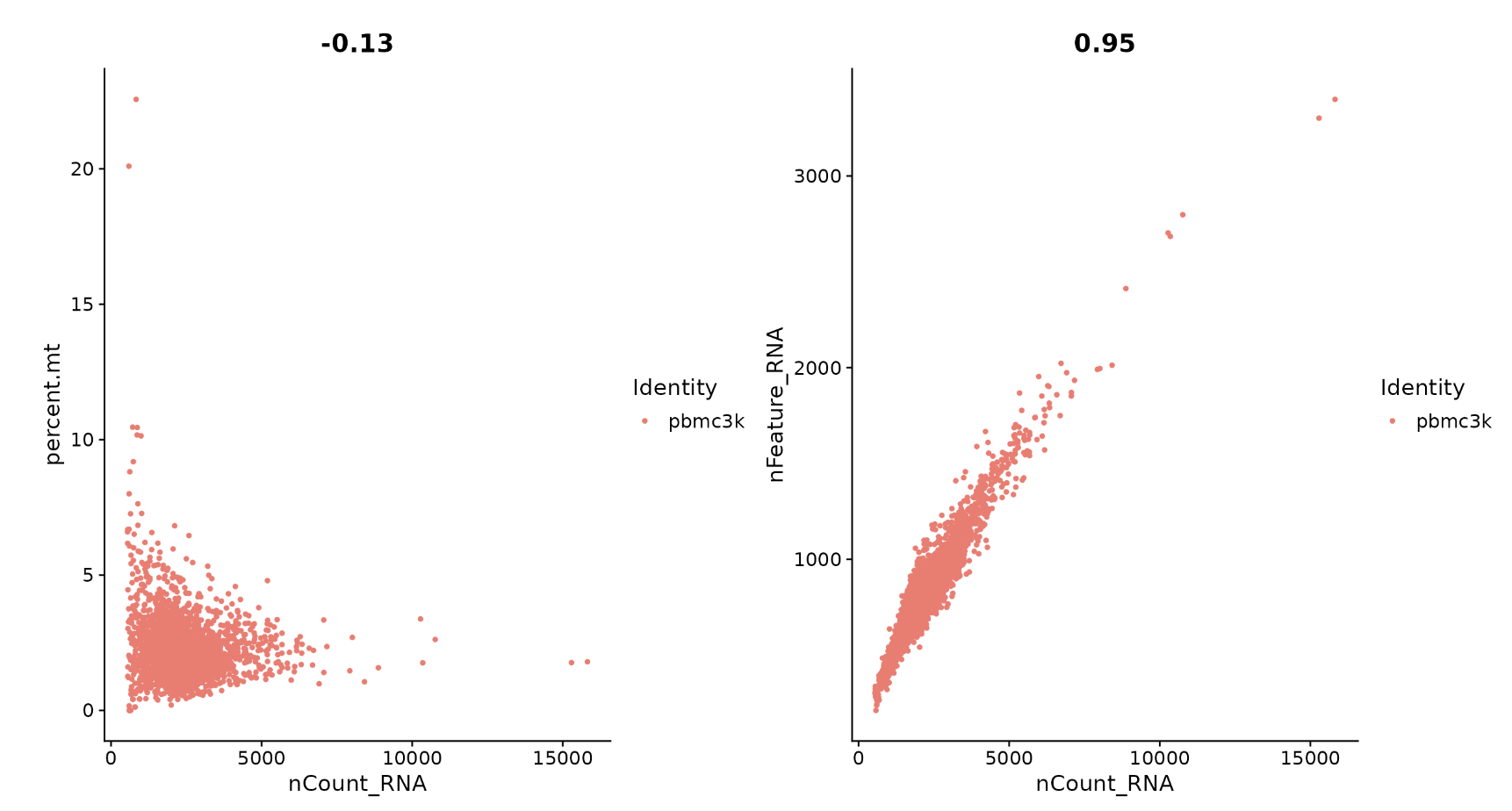
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
未完待续,持续关注!
Source: https://zenghensatijalab.org/seurat/articles/pbmc3k_tutorial
[2]data: https://cf.10xgenomics.com/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz
本文由 mdnice 多平台发布
这篇关于Seurat - 聚类教程 (1)的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!







