本文主要是介绍单细胞seurat入门—— 从原始数据到表达矩阵,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
根据所使用的建库方法,单细胞的RNA序列(也称为读取(reads)或标签(tags))将从转录本的3'端(或5'端)(10X Genomics,CEL-seq2,Drop-seq,inDrops)或全长转录本(Smart-seq)获得。

图片来源: Papalexi E and Satija R. Single-cell RNA sequencing to explore immune cell heterogeneity, Nature Reviews Immunology 2018 (https://doi.org/10.1038/nri.2017.76)
我们可以根据自己感兴趣的生物学问题而选择不同的方法。这些方法具有以下优点:
- 3'(或5')端测序:
- 通过独特的分子标记物(molecular identifiers)来更准确地定量鉴别生物复制品和扩增(PCR)复制品
- 可以给更多的细胞测序,更好地识别细胞类群
- 每个细胞的平均测序成本更低
- 最适用于10000个以上的细胞
- 全长(Full length)测序
- 可以检测到亚型水平上的表达差异
- 可以进行等位基因(allele-specific)表达差异的检测
- 可以给少量细胞进行更深度的测序
- 最适用于细胞量少的样本
全长测序和3'端测序需要进行许多相同的分析步骤,但3'端流程越来越受欢迎,在分析过程中包含了更多的步骤。因此,我们的教程将详细分析这些3'端流程的数据,重点是基于液滴的方法(inDrops,Drop seq,10X Genomics)。
3’端测序(包括所有基于液滴的方法)
对于单细胞RNA测序的数据分析来说,理解在每次读取中获得的信息,以及我们如何在分析过程中使用这些信息是非常有帮助的。
对于3'端测序的方法,来自同一转录本不同分子的读取只能来自转录本的3'端,因此具有相同序列的可能性很高。然而,在建库过程中的PCR步骤也可能产生读取重复。为了确定一次读取是生物重复还是技术重复,这些方法使用了唯一的分子标识符(UMIs)。
- 使用不同UMI映射到同一个转录本的读取来自不同的分子,是生物学上的复制-每个读取都应该被计数。
- 具有相同UMI的读取源于同一分子,是技术上的复制-这些UMI应该合并为一个读取的计数。
- 在下图中,ACTB的读取应合并计为单个读取,而ARL1的读取应分别计为单个读取。

图片来源: modified from Macosko EZ et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets, Cell 2015 (https://doi.org/10.1016/j.cell.2015.05.002)
所以我们需要追踪UMI,除此之外,我们还需要什么信息来正确量化样本中每个细胞中每个基因的表达呢?无论使用何种基于液滴的方法,在细胞水平上进行正确的定量需要以下条件:
- 样本索引 (Sample index):确定读取来自于哪个样本。在建库期间添加——需要记录
- 细胞条形码 (Celluar barcode):确定读取来自于哪个细胞。每个建库方法都有一个供在建库期间使用的细胞条形码库存(stock)
- 唯一分子标识符 (UMI):确定读取来自哪个转录分子。UMI将被用于合并PCR复制物
- 测序读取1 (Sequencing read1):1号读取序列
- 测序读取2 (Sequencing read2):2号读取序列
例如,当使用inDrops v3建库方法时,以下内容显示如何在四次读取中获取所有信息:

图片来源:Sarah Boswell, Director of the Single Cell Sequencing Core at HMS
- R1 (61bp 读取1):读取的序列(顶部红色箭头)
- R2 (8bp 索引读取1(i7)):细胞条码 —— 判断读取来自于哪个细胞(顶部紫色箭头)
- R3 (8bp 索引读取2(i5)):样本/库索引 —— 判断读取来自哪个样本(底部红色箭头)
- R4 (14bp 读取2):读取2和剩下的细胞条形码和UMI —— 读取来自于哪个转录本(底部紫色箭头)
对于不同的基于液滴的单细胞RNA测序方法,分析流程是相似的,但是对UMI、细胞ID和样本索引的分析将有所不同。例如,下面是10X序列读取的示意图,其中索引、UMI和条码的位置不同:

图片来源: Sarah Boswell, Director of the Single Cell Sequencing Core at HMS
单细胞RNA测序的工作流程
单细胞RNA测序方法将确定如何从序列读取中解析条码和UMI。因此,尽管一些具体步骤略有不同,总体工作流程通常会遵循相同的步骤,而不考虑方法的差异。一般工作流程如下:

图片来源: Luecken, MD and Theis, FJ. Current best practices in single‐cell RNA‐seq analysis: a tutorial, Mol Syst Biol 2019 (doi: https://doi.org/10.15252/msb.20188746)
工作流程的步骤是:
- 生成计数矩阵 (Generation of the count matrix):格式化读取、分离样本、映射(mapping)和定量(quantification)
- 原始计数矩阵的质量控制 (Quality control of the raw counts):过滤掉质量差的细胞
- 过滤后计数的聚类 (Clustering of filtered counts):将转录活动相似的细胞归为一类(细胞类型=不同的聚类)
- 标记识别 (Marker identification):识别每个细胞群的基因标记(marker)
- 其他可选的下游步骤 (Optional downstream steps)
不管做什么样的分析,基于每种情况的单一样本而得出关于群体的结论都是不可信的。生物学重复仍然是非常必要的!也就是说,如果你想得出与总体相对应的结论,不应该局限于单一样本。
生成计数矩阵
我们将从讨论该工作流程的第一部分开始,该部分是从原始序列数据生成计数矩阵。我们将重点关注基于液滴的3'端测序,如inDrops、10X Genomics和Drop seq。

测序后,测序设备将原始测序数据输出为BCL或FASTQ格式,或生成计数矩阵 (count matrix)。如果读取的是BCL格式,那么我们需要转换为FASTQ格式。有一个名为bcl2fastq的命令行工具可以轻松地完成此转换。
注:在工作流程的这一步,我们不进行分离。您可能已经对6个样本进行了测序,但所有样本的读取可能都出现在同一个BCL或FASTQ文件中。
在许多单细胞RNA测序方法中,从原始数据生成计数矩阵将会经历很多类似的步骤。

umis 和 zUMIs 是用来估算3'端转录本测序数据表达量的命令行工具。两种工具都包含了UMIs的合并以校正扩增偏差(amplification bias)的功能。此过程中的步骤包括:
- 格式化读写并过滤低质量的细胞条码
- 分离样本
- 比对/伪映射到转录组
- 合并UMI并量化读写
如果使用10X Genomic建库方法, Cell Ranger 流程将会被用于以上所有的步骤。
1. 格式化读写并过滤低质量的细胞条码
FASTQ文件可被用于解析细胞条码、UMI和样本条码。对于基于液滴的方法,由于以下原因,许多细胞条码将会匹配到数量较少(< 1000)的读取,这是因为:
- 封装了来自于死/濒死细胞的游离RNA
- 混入了只表达少量基因的简单细胞(如红细胞等)
- 其他因素的影响
这些多余的条码需要在读取比对之前从测序数据中过滤掉。要进行此筛选,将提取并保存每个细胞的“细胞条码”和“分子条码”。例如,如果使用了umis工具,每次读取时都会将信息添加到标题行,格式如下:
@HWI-ST808:130:H0B8YADXX:1:1101:2088:2222:CELL_GGTCCA:UMI_CCCTAGGAAGATGGAGGAGAGAAGGCGGTGAAAGAGACCTGTAAAAAGCCACCGN+@@@DDBD>=AFCF+<CAFHDECII:DGGGHGIGGIIIEHGIIIGIIDHII#建库方法中使用的细胞条码应该是已知的,且未知的条码将会被丢弃,同时允许存在适量与已知细胞条码不匹配的现象。
2. 分离样本读取
如果测序不止一个样本,这个过程的下一步是对样本进行分离。这个步骤不是由umis工具完成,而是由zUMIs完成的。我们需要对读取的数据进行分析,以确定与每个细胞关联的样本条码。
3. 映射/伪映射至cDNA
为了确定读取源于哪个基因,可使用传统的(STAR)或轻量级方法(Kallisto/RapMap)对读取进行映射 (mapping)。
4. 合并UMI并完成对读取的定量
重复的UMI被合并,这样唯一的UMI可以使用Kallisto或featureCounts这样的工具定量。结果是一个细胞的基因计数矩阵:
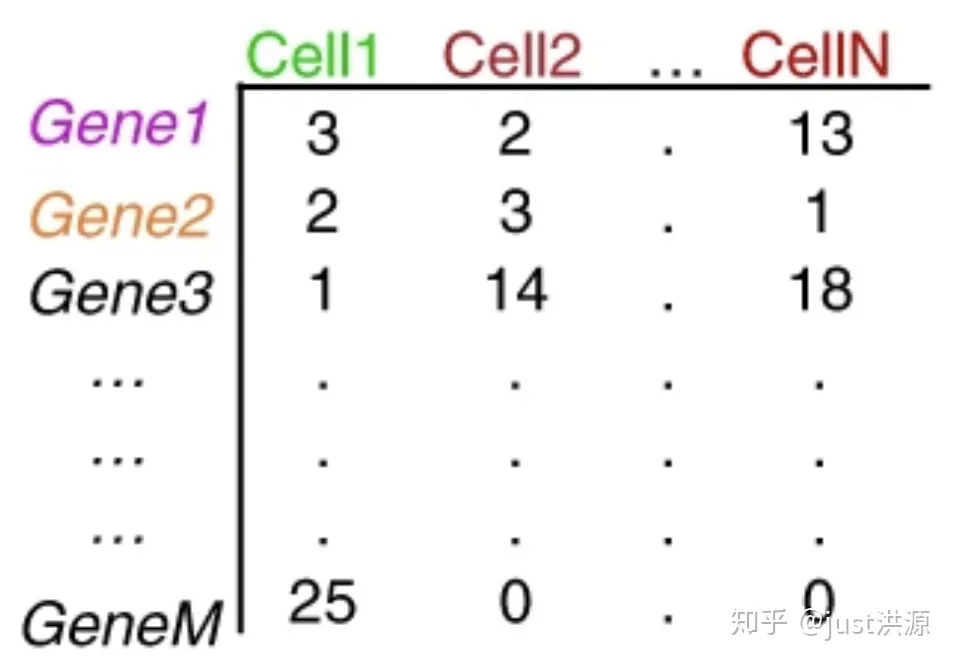
图片来源:extracted from Lafzi et al. Tutorial: guidelines for the experimental design of single-cell RNA sequencing studies, Nature Protocols 2018 (https://doi.org/10.1038/s41596-018-0073-y)
矩阵中的每个值表示一个细胞中相应基因的读取数。使用计数矩阵,我们可以探索和过滤数据,只保留较高质量的细胞。
这篇关于单细胞seurat入门—— 从原始数据到表达矩阵的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!







