本文主要是介绍单细胞实战(1)数据下载-数据读取-seurat对象创建,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
这篇文章我们将介绍从geo数据库下载单细胞测序数据后,多种数据格式多样本情况下,如何读取数据并创建seurat对象。
本文主要结构:
一、数据下载
二、数据读取与seurat对象创建
- 单样本情况下各种格式数据的读取,读取后seurat对象的创建
- 多样本情况下各种格式数据的读取,读取后seurat对象的创建、合并
一、数据下载
大家自行去GEO官网(https://www.ncbi.nlm.nih.gov/gds)搜索下载自己想要的单细胞测序数据。本文后面会提供数据用于示例代码测试。
GEO数据库上提供的单细胞测序数据常见格式主要有以下几种:
-
10x Genomics格式: matrix.mtx、genes.tsv和barcodes.tsv文件是10X Genomics单细胞转录组测序数据的标准文件格式。这些文件通常存储在一个目录中,可以使用Read10X函数从R语言中读取。
- matrix.mtx:这是一个稀疏矩阵文件,其中包含了每个单细胞的基因表达信息。矩阵中的每一行代表一个基因,每一列代表一个单细胞,矩阵中的每个元素表示该基因在该单细胞中的表达量。
- genes.tsv(或features.tsv):这是一个文本文件,其中包含了每个基因的信息。每一行代表一个基因,每一列代表一个属性,例如基因名称、基因编号等。
- barcodes.tsv:这是一个文本文件,其中包含了每个单细胞的条形码信息。每一行代表一个单细胞,每一列代表一个属性,例如条形码序列、细胞类型等。
-
h5格式: 这是一种用于存储大规模数据的二进制文件格式,它可以包含多种数据类型,如矩阵、表格、图像等。
-
压缩文本矩阵(TXT或CSV的GZ文件): 压缩文本矩阵可以用于存储单细胞测序数据的表达矩阵或元数据,它可以减少文件的大小和传输时间 。
-
h5ad格式: 它专门用于存储和分享单细胞表达数据,它使用Anndata库来创建和读取。h5ad格式可以与cellxgene或Seurat等工具兼容,进行单细胞数据的可视化和分析 。
-
h5seurat格式: 这是一种基于h5格式的文件格式,它专门用于存储和分析多模态单细胞和空间分辨率表达实验,如CITE-seq或10X Visium等技术。h5seurat格式可以与SeuratDisk等工具兼容,进行单细胞数据的读写 。
-
R数据文件(RDS/RDATA文件): 以R语言的数据文件格式存储表达式矩阵,需要R软件直接读取。
二、数据读取与seurat对象创建
单样本
单样本情况下每种格式的数据读取与seurat对象创建演示:
10x Genomics格式:
演示数据的下载:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE234527
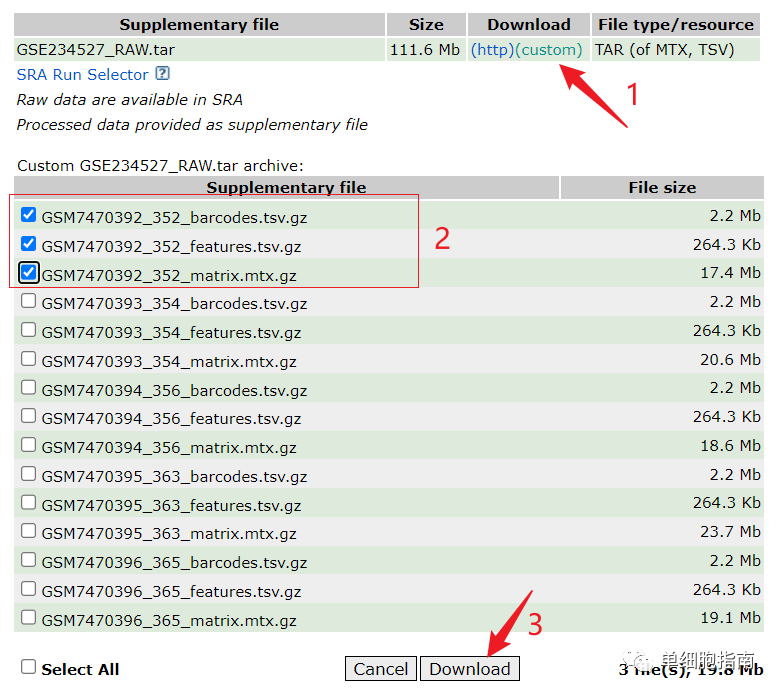
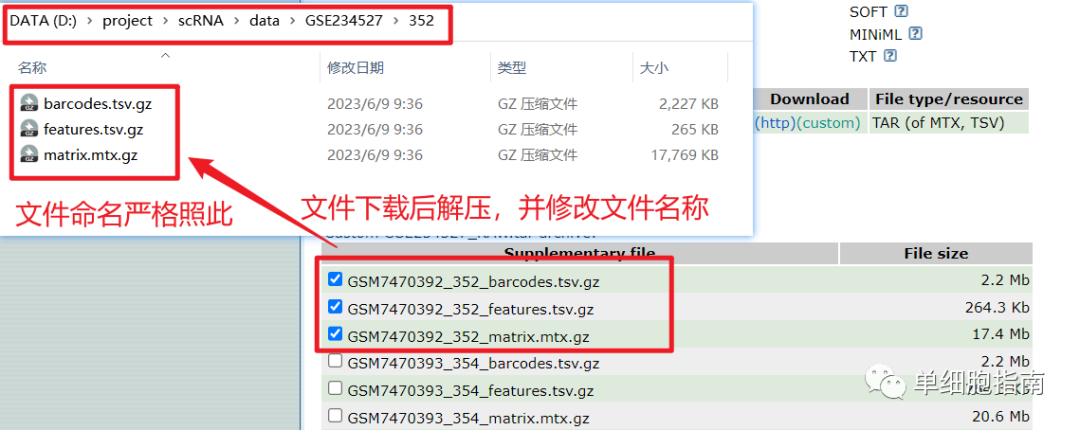
文件下载后解压,并修改名称,存放路径如下图:
读取文件并创建对象的代码参考:
# 导入Seurat包
library(Seurat)# 查看当前工作目录
getwd()# 设置工作目录(将工作目录切换到指定路径下)
setwd("D:/project/scRNA")# 读取10x数据,data.dir参数指定存放文件的路径
seurat_data <- Read10X(data.dir = "./data/GSE234527/352")# 创建Seurat对象
seurat_obj <- CreateSeuratObject(counts = seurat_data,project = "GSM7470392_352",min.features = 200,min.cells = 3)# 查看Seurat对象的基本信息
seurat_obj
h5格式:
演示数据的下载:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE200874
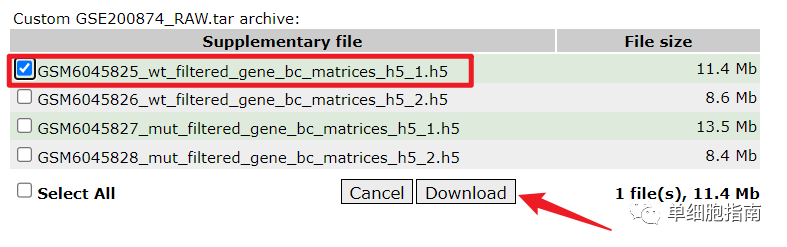
下载后解压,存放路径如图
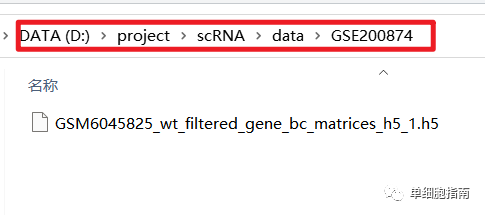
读取文件并创建对象的代码参考:
# 导入Seurat包
library(Seurat)# 查看当前工作目录
getwd()# 设置工作目录(将工作目录切换到指定路径下)
setwd("D:/project/scRNA")# 指定要读取的文件所在位置和文件名称
h5_file <- "./data/GSE200874/GSM6045825_wt_filtered_gene_bc_matrices_h5_1.h5"# 读取h5格式的文件(使用Read10X_h5函数读取h5格式的单细胞数据文件)
seurat_data <- Read10X_h5(file = h5_file)# 创建Seurat对象(使用CreateSeuratObject函数创建Seurat对象,并将读取的h5格式数据转换为Seurat对象)
seurat_obj <- CreateSeuratObject(counts = seurat_data,project = "GSM6045825_wt",min.features = 200,min.cells = 3)# 查看Seurat对象的基本信息
seurat_obj
压缩文本矩阵(TXT或CSV的GZ文件):
这两种文件建议先手动加压到本地查看一下文件内容格式。
CSV压缩GZ格式演示数据的下载:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gse130148
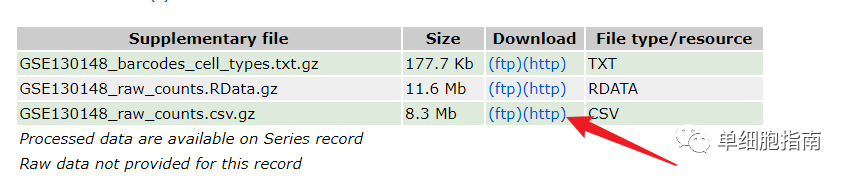
下载后文件的存放路径
CSV压缩GZ格式示例代码:
# 导入Seurat包
library(Seurat)# 查看当前工作目录
getwd()# 设置工作目录(将工作目录切换到指定路径下)
setwd("D:/project/scRNA")# 使用read.csv()函数从csv.gz格式的文件中读取数据,并将第一列作为行名
seurat_data<- read.csv(gzfile("./data/GSE130148/GSE130148_raw_counts.csv.gz"), row.names = 1)# 使用CreateSeuratObject()函数创建Seurat对象,并在此处指定项目名称
seurat_obj <- CreateSeuratObject(counts = seurat_data,min.features = 200,min.cells = 3, project = "GSE130148")
txt压缩GZ格式示例代码:
# 导入Seurat包
library(Seurat)# 查看当前工作目录
getwd()# 设置工作目录(将工作目录切换到指定路径下)
setwd("D:/project/scRNA")# 使用read.table()函数从txt.gz格式的文件中读取数据,并将第一列作为行名
seurat_data<- read.table(gzfile("./data/GSE130xxx/xxxx.txt.gz"), row.names = 1, header = TRUE, sep = "\t")# 使用CreateSeuratObject()函数创建Seurat对象,并在此处指定项目名称
seurat_obj <- CreateSeuratObject(counts = seurat_data,min.features = 200,min.cells = 3, project = "GSE130xxx")
h5ad格式:
下载测试文件:
https://www.dropbox.com/s/ngs3p8n2i8y33hj/pbmc3k.h5ad?dl=0
# 下载测试文件
# https://www.dropbox.com/s/ngs3p8n2i8y33hj/pbmc3k.h5ad?dl=0# 导入所需的R包
library(Seurat)
# 安装SeuratDisk包
#remotes::install_github("mojaveazure/seurat-disk")
library(SeuratDisk)# 查看当前工作目录
getwd()# 设置工作目录(将工作目录切换到指定路径下)
setwd("D:/project/scRNA")# 将h5ad格式文件转换为h5seurat格式文件,同时指定使用的assay为"RNA"
Convert("./data/pbmc/pbmc3k.h5ad", "h5seurat", overwrite = TRUE, assay = "RNA")# 使用LoadH5Seurat()函数加载h5seurat格式文件,并创建Seurat对象
seurat_pbmc <- LoadH5Seurat("./data/pbmc/pbmc3k.h5seurat")
R数据文件(RDS/RDATA文件)
# 使用load()函数读取RDATA文件
load("path/to/your/file.Rdata")# 使用readRDS()函数读取RDS文件
my_data <- readRDS("path/to/your/file.rds")
多样本
多样本情况下我们主要关注10x Genomics格式和压缩文本矩阵(TXT或CSV的GZ文件)
10x Genomics格式多样本读取与对象创建:
测试数据下载:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE234527
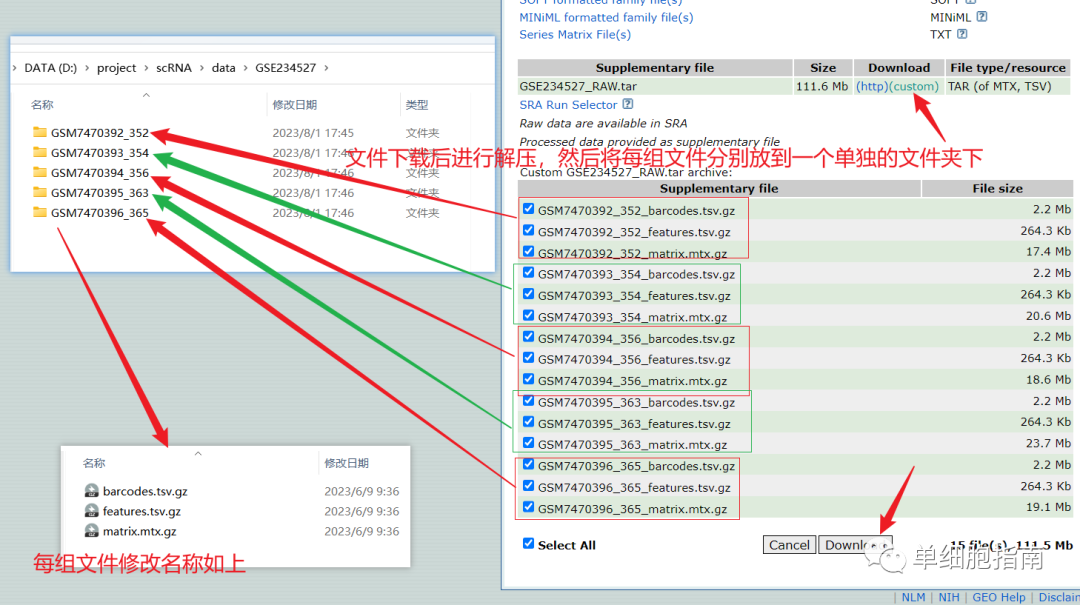
示例代码:
# 导入Seurat包
library(Seurat)# 查看当前工作目录
getwd()# 设置工作目录(将工作目录切换到指定路径下)
setwd("D:/project/scRNA")# 获取数据文件夹下的所有样本文件列表
samples <- list.files("./data/GSE234527")# 创建一个空的列表来存储Seurat对象
seurat_list <- list()# 读取每个样本的10x数据并创建Seurat对象
for (sample in samples) {
# 拼接文件路径data.path <- paste0("./data/GSE234527/", sample)# 读取10x数据,data.dir参数指定存放文件的路径seurat_data <- Read10X(data.dir = data.path)# 创建Seurat对象,并指定项目名称为样本文件名seurat_obj <- CreateSeuratObject(counts = seurat_data,project = sample,min.features = 200,min.cells = 3)# 将Seurat对象添加到列表中seurat_list <- append(seurat_list, seurat_obj)
}# 打印所有的Seurat对象列表
seurat_list# 合并Seurat对象,将所有Seurat对象合并到一个对象中
seurat_combined <- merge(seurat_list[[1]], y = seurat_list[-1],add.cell.ids = samples)
# 打印合并后的Seurat对象
print(seurat_combined)
h5格式多样本数据读入与对象创建:
测试数据下载:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE200874
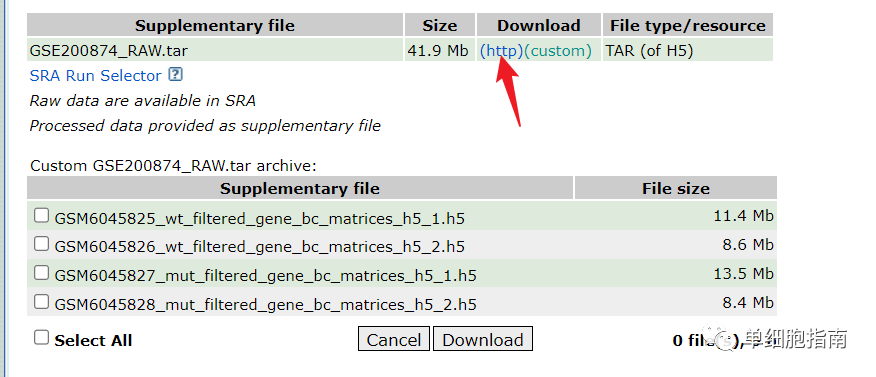
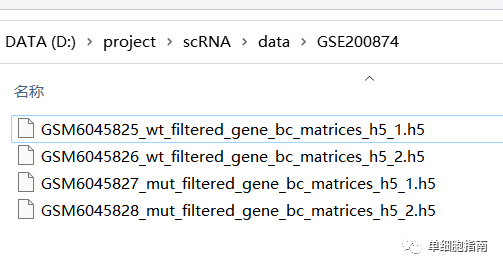
下载后将数据解压:
# 导入Seurat包
library(Seurat)# 设置工作目录
setwd("D:/project/scRNA")# 获取数据文件夹下的所有h5文件列表
h5_files <- list.files("./data/GSE200874", pattern = "\\.h5$")# 创建一个空的列表来存储Seurat对象
seurat_list <- list()# 循环读取每个h5文件的数据并创建Seurat对象
for (h5_file in h5_files) {# 拼接文件路径data.path <- paste0("./data/GSE200874/", h5_file)# 读取h5数据seurat_data <- Read10X_h5(filename = data.path)# 创建Seurat对象,并指定项目名称为文件名sample_name <- tools::file_path_sans_ext(basename(h5_file))seurat_obj <- CreateSeuratObject(counts = seurat_data,project = sample_name,min.features = 200,min.cells = 3)# 将Seurat对象添加到列表中seurat_list <- append(seurat_list, seurat_obj)
}# 提取下划线前面的部分
sample_names <- sub("_.*", "", h5_files)
# 合并Seurat对象,将所有Seurat对象合并到一个对象中
seurat_combined <- merge(seurat_list[[1]],y = seurat_list[-1],add.cell.ids = sample_names)
# 打印合并后的Seurat对象
print(seurat_combined)
压缩文本矩阵(TXT或CSV的GZ文件)多样本:
下载测试文件:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi
下载三个数据演示一下代码
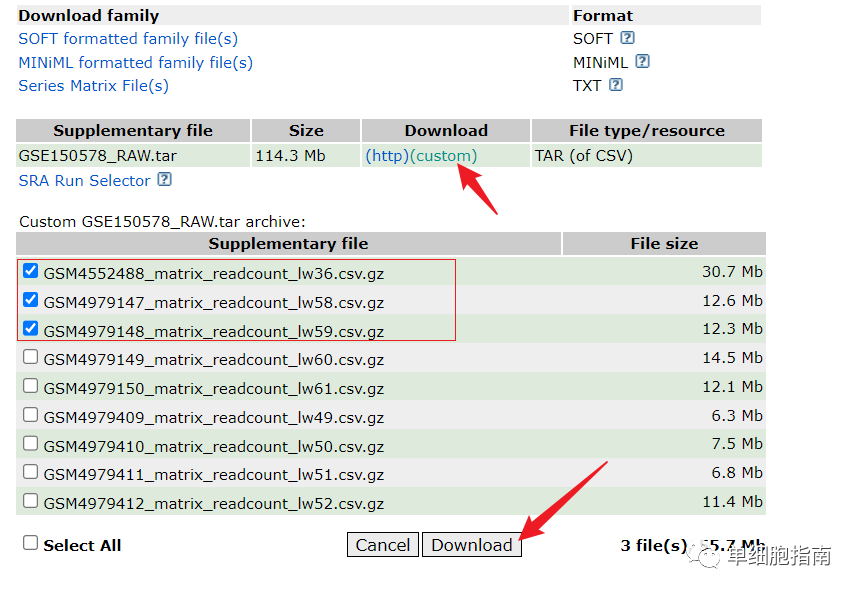
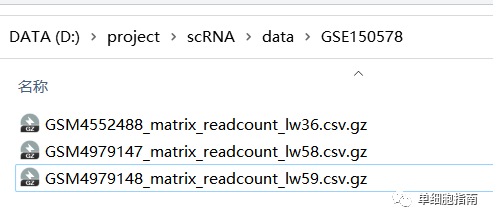
下载后解压:
示例代码:
# 导入Seurat包
library(Seurat)# 导入Seurat包
library(Seurat)# 设置工作目录为存放数据文件的路径
setwd("D:/project/scRNA")# 获取所有csv.gz文件的列表
file_list <- list.files("./data/GSE150578", pattern = "\\.csv\\.gz$")# 创建一个空的列表来存储Seurat对象
seurat_list <- list()# 循环读取每个csv.gz文件的数据并创建Seurat对象
for (file in file_list) {# 拼接文件路径data.path <- paste0("./data/GSE150578/", file)# 读取csv.gz文件数据data <- read.csv(gzfile(data.path), row.names = 1)# 创建Seurat对象,并指定项目名称为文件名(去除后缀)sample_name <- tools::file_path_sans_ext(basename(file))seurat_obj <- CreateSeuratObject(counts = seurat_data,project = sample_name,min.features = 200,min.cells = 3)# 将Seurat对象添加到列表中seurat_list <- append(seurat_list, seurat_obj)
}# 提取下划线前面的部分
sample_names <- sub("_.*", "", file_list)
# 合并Seurat对象,将所有Seurat对象合并到一个对象中
seurat_combined <- merge(seurat_list[[1]],y = seurat_list[-1],add.cell.ids = sample_names)
# 打印合并后的Seurat对象
print(seurat_combined)
大家可以使用fread()等更高效的函数代替文中的read.csv()函数,但是要注意读取后数据格式是否准确。
参考链接:https://www.jianshu.com/p/5b26d7bc37b7
参考链接:https://mp.weixin.qq.com/s/M15kWdH8eDONfakNhY-enA
这篇关于单细胞实战(1)数据下载-数据读取-seurat对象创建的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!






