seq专题

使用seq_file
在《使用procfs》一文的源码示例中有说到proc文件系统每次读取的数据只能是1个页,如果超过则需多次读取,这样的话会增加读取次数,增多系统调用次数,影响了整体的效率,故而才有seq file序列文件的出现,该项功能使得内核对于大文件的读取更加容易。 对于seq file,其结构体定义在include/linux/seq_file.h文件中,内容如下: struct seq_file {

【0323】Postgres内核之 hash table sequentially search(seq_scan_tables、num_seq_scans)
0. seq scan tracking 我们在这里跟踪活跃的 hash_seq_search() 扫描。 需要这种机制是因为如果扫描正在进行时发生桶分裂(bucket split),它可能会访问两次相同的条目,甚至完全错过某些条目(如果它正在访问同一个分裂的桶中的条目)。因此,如果正在向表中插入数据,我们希望抑制桶分裂。 在当前的使用中,这种情况非常罕见,因此只需将分裂推迟到下一次插入即可。

2024.09.04【读书笔记】|如何使用Tombo进行Nanopore Direct RNA-seq(DRS)分析
文章目录 Tombo快速使用介绍模型介绍RNA修饰分析步骤特异性替代碱基检测(推荐)De novo canonical model comparison ONT全长转录组分析步骤疑难解答Minimap2在比对nanopore直接RNA-seq数据时的最佳实践和参数设置有哪些?featureCounts在进行RNA-seq定量分析时,如何选择最合适的参考基因组注释文件?Tombo序列重校正过程

关于宏CV_GET_SEQ_ELEM
#define CV_GET_SEQ_ELEM(TYPE,seq,index)\(TYPE*)cvGetSeqElem((CvSeq*)(seq),(index)) 用法:从所给序列中取出元素的地址,注意:得到的是地址,即指针 所以,关键在于序列中存放的是那种类型的数据,若存放的为地址,那用这个宏得到的就是指针的指针。 例程: 1.序列中存放的为CvPoint CvPoint pt=*C

ChIP-seq项目文章 | Adv Sci转录因子FOXK1通过驱动小管上皮细胞糖酵解促进慢性肾脏疾病
在慢性肾脏疾病(CKD)进展过程中,肾小管上皮细胞(TECs)经历了从脂肪酸氧化到糖酵解的能量相关代谢转变。然而,这种糖酵解爆发的机制尚不清楚。2024年7月31日,武汉大学王惠明教授团队和湖北民族大学刘伦志教授团队在Advanced Science(IF:14.6)上在线发表了题为“Forkhead Box Protein K1 Promotes Chronic Kidney Disease b
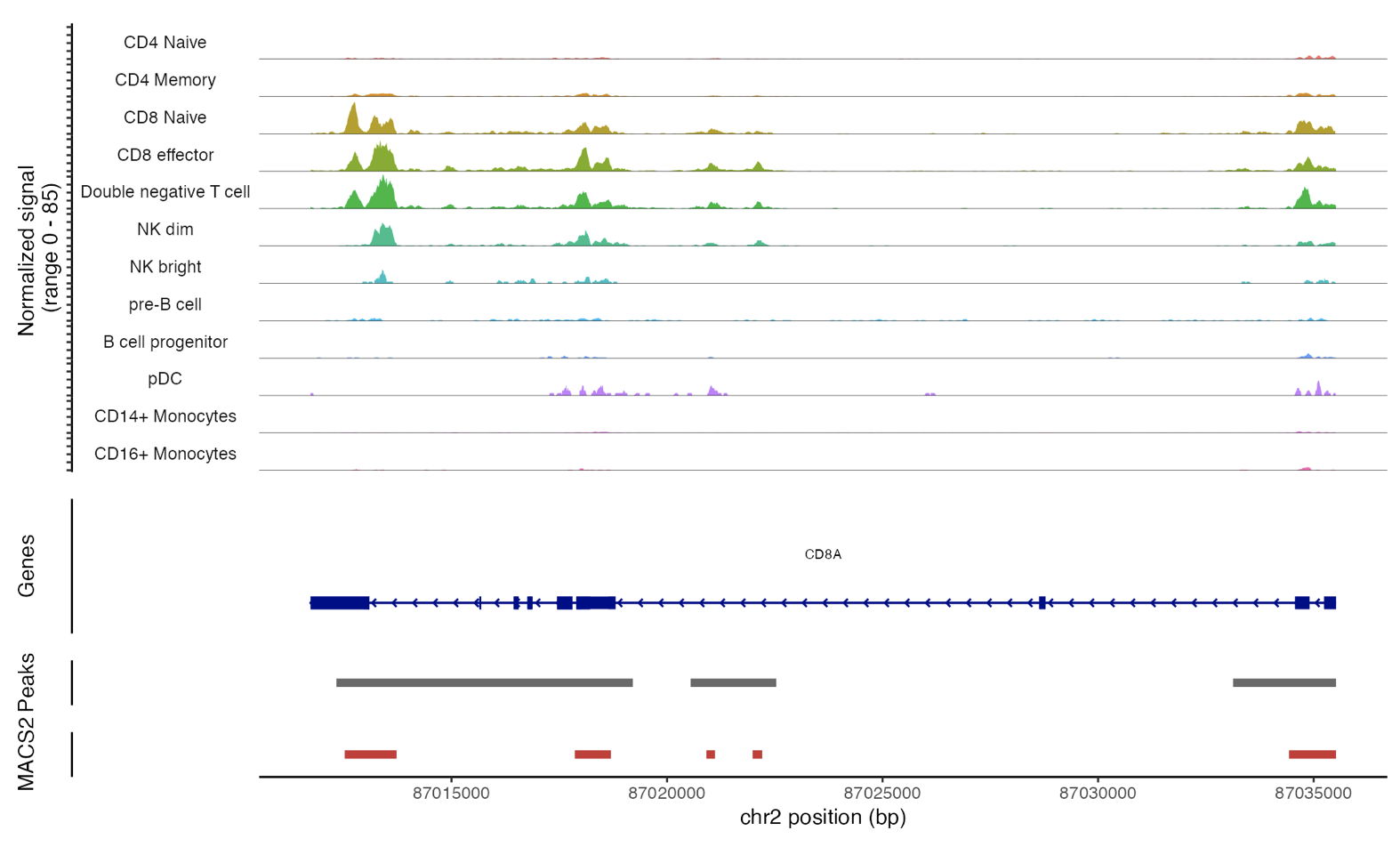
Signac 单细胞|ATAC-seq Call peak
引言 本文将向您展示如何利用MACS2软件,在单细胞ATAC-seq的基因组数据中识别基因调控区域的峰值。 实战 在使用Signac进行峰值检测之前,您需要先安装MACS2。您可以通过pip或conda安装它,或者从源代码自行编译。 本次演示以人类外周血单核细胞的单细胞ATAC-seq数据为例。首先,请加载必要的软件包和预先处理过的Seurat数据对象。 library(Signac)libra

使用stack分析RAD-seq
一次简化基因组数据分析实战 尽管目前已经有大量物种基因组释放出来,但还是存在许多物种是没有参考基因组。使用基于酶切的二代测序技术,如RAD-seq,GBS,构建遗传图谱是研究无参考物种比较常用的方法。Stacks就是目前比较通用的分析流程,能用来构建遗传图谱,处理群体遗传学,构建进化发育树。 这篇教程主要介绍如何使用Stacks分析基于酶切的二代测序结果,比如说等RAD-seq,分析步骤为环境

使用SCALE分析单细胞ATAC-seq数据
SCALE全称是Single-Cell ATAC-seq analysis vie Latent feature Extraction, 从名字中就能知道这个软件是通过隐特征提取的方式分析单细胞ATAC-seq数据。 在文章中,作者从开发者的角度列出了目前的scATAC-seq分析软件,chromVAR, scABC, cisTopic, scVI,发现每个软件都有一定的不足之处,而从我们软件使
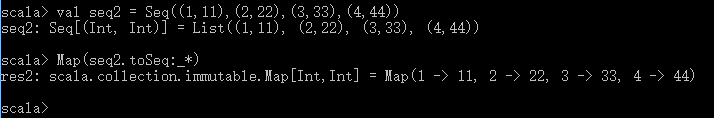
Scala中Seq转Map的方法(:_*)
近日在学习Kafka源码的时候,对代码中 .toSeq: _*的语法不是很理解,于是在scala shell中做了几次尝试,理解了其中的用法含义。 1. 源码 topicRegistry的数据结构: 2. 尝试 1)Seq(1,2,3,4) 回到源码查看topicRegistry的数据结构,发现调用toSeq方法的数据结构是元组的List,而且语法:_*是在Map()构造函数
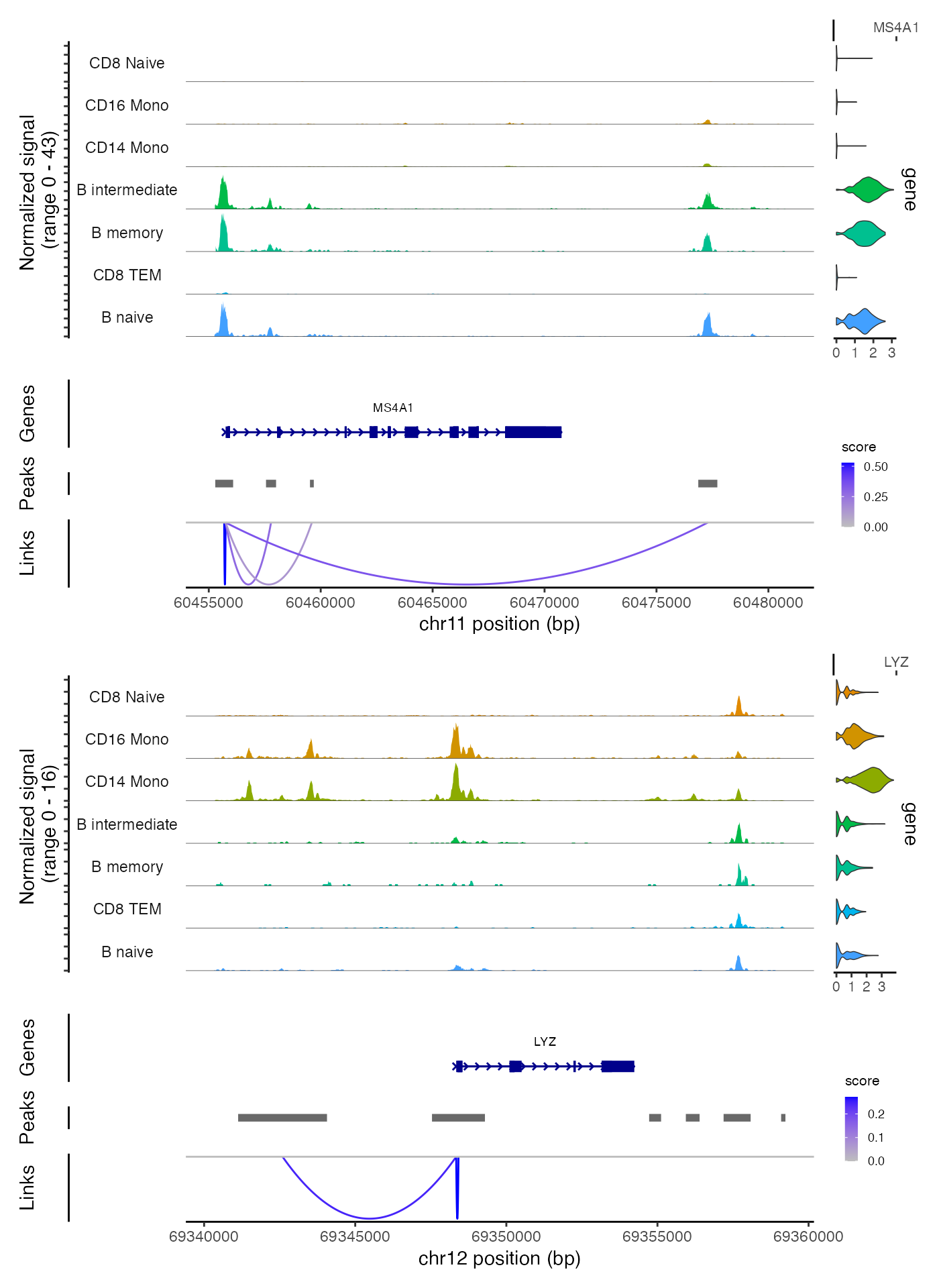
单细胞|RNA-seq ATAC-seq 联合分析
引言 本文[1]将介绍如何利用Signac和Seurat这两个工具,对一个同时记录了DNA可接触性和基因表达的单细胞数据集进行综合分析。我们将以一个公开的10x Genomics Multiome数据集为例,该数据集针对的是人体的外周血单核细胞。 数据准备 library(Signac)library(Seurat)library(EnsDb.Hsapiens.v86)library(BSgen
项目文章 | Cell ReportsChIP-seq和RNA-seq联合鉴定伯克霍尔德氏菌毒性的重要调节因子
发表单位:中山大学深圳校区制药科学学院 发表日期:2024年5月14日 研究期刊:Cell Reports(IF: 8.8) 研究材料:伯克霍尔德氏菌 主要技术:ChIP-seq,EMSA,微尺度热泳分析,RNA-seq, RT-qPCR 近日,中山大学深圳校区制药科学学院邓音乐教授研究团队在Cell Reports上发表了题为“Regulation of Burkholderi
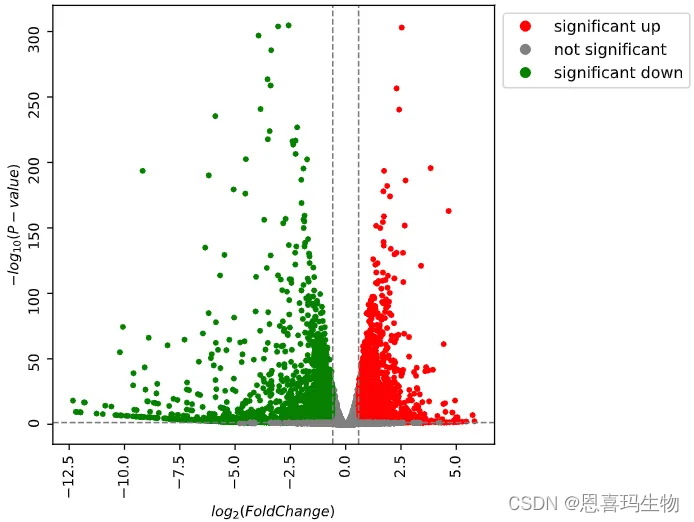
RNA-seq上下游分析snakemake流程
学习完snakemake后写的第一个流程是RNA-seq上游定量和下游的质控和差异分析。 使用fastp处理fastq文件,在使用START比对到基因组同时得到raw count,使用非冗余外显子长度作为基因的长度计算FPKM、TPM,同时也生成了CPM的结果。 非冗余外显子长度计算可以参考之前的推文转录组实战02: 计算非冗余外显子长度之和 对定量结果质控使用生信技能树的三张图(PCA、树

inferCNV:scRNA-seq数据推断染色体拷贝数变化
inferCNV分析简介 inferCNV用于探索肿瘤单细胞RNA-Seq 数据,以确定体细胞大规模染色体拷贝数改变的证据,例如整个染色体或大片段染色体的增益或缺失。这是通过与一组参考“正常”细胞(这里的正常细胞可自行定义)进行比较,探索肿瘤基因组各部位基因的表达强度来完成的。热图展示每个染色体的相对表达强度,并且与“正常”细胞相比,肿瘤基因组的哪些区域过表达或降低(异常)。 同时我们需要了解
易基因:RNA免疫共沉淀测序 (RIP-seq) 技术介绍
大家好,这里是专注表观组学十余年,领跑多组学科研服务的易基因。 RIP-seq是将RNA免疫共沉淀(RNA Immunoprecipitation,RIP)与二代测序技术(NGS)相结合以研究细胞内RNA与蛋白互作的技术,RIP利用目标蛋白抗体把相应的RNA-蛋白复合物(RNA Binding Protein,RBP)沉淀下来,然后经过富集和纯化就可以对结合在复合物上的RNA进行测序分析。 R
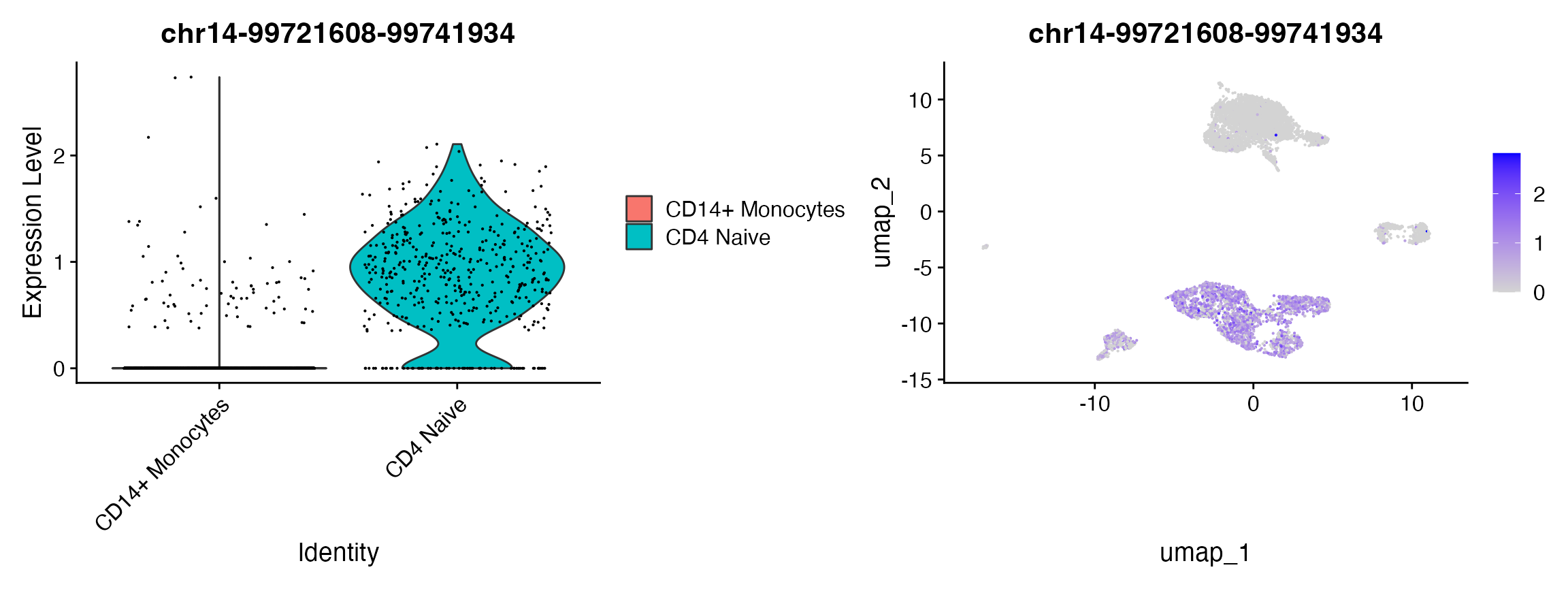
单细胞分析(Signac): PBMC scATAC-seq 整合
引言 在本教学指南中,我们将探讨由10x Genomics公司提供的人类外周血单核细胞(PBMCs)的单细胞ATAC-seq数据集。 加载包 首先加载 Signac、Seurat 和我们将用于分析人类数据的其他一些包。 if (!requireNamespace("EnsDb.Hsapiens.v75", quietly = TRUE)) BiocManager::install("Ens
Oracle 序列使用时:ORA-08002: 序列 SEQ_WGB_TEST2.CURRVAL 尚未在此会话中定义
一、场景:Oracle中id经常使用序列自增,这就会导致新增时id的使用(A表的主键ID,新增时,同时要在B表中存入一个相同的当前ID) 二、使用:CURRVAL(当前值) NEXTVAL(下一个值): NEXTVAL可以单独使用在sql语句中;而CURRVAL在没有使用NEXTVAL的时候使用的话就会报错(尚未在此会话中定义)。 三、解决:在执行CURRVAL之前需要先执行NEXTVAL
scala基础----序列trait:Seq、IndexedSeq及LinearSeq
Seq trait用于表示序列。所谓序列,指的是一类具有一定长度的可迭代访问的对象,其中每个元素均带有一个从0开始计数的固定索引位置。 序列的操作有以下几种,如下表所示: 索引和长度的操作 apply、isDefinedAt、length、indices,及lengthCompare。序列的apply操作用于索引访问;因此,Seq[T]类型的序列也是一个以单个Int(索引下标)为参数、返
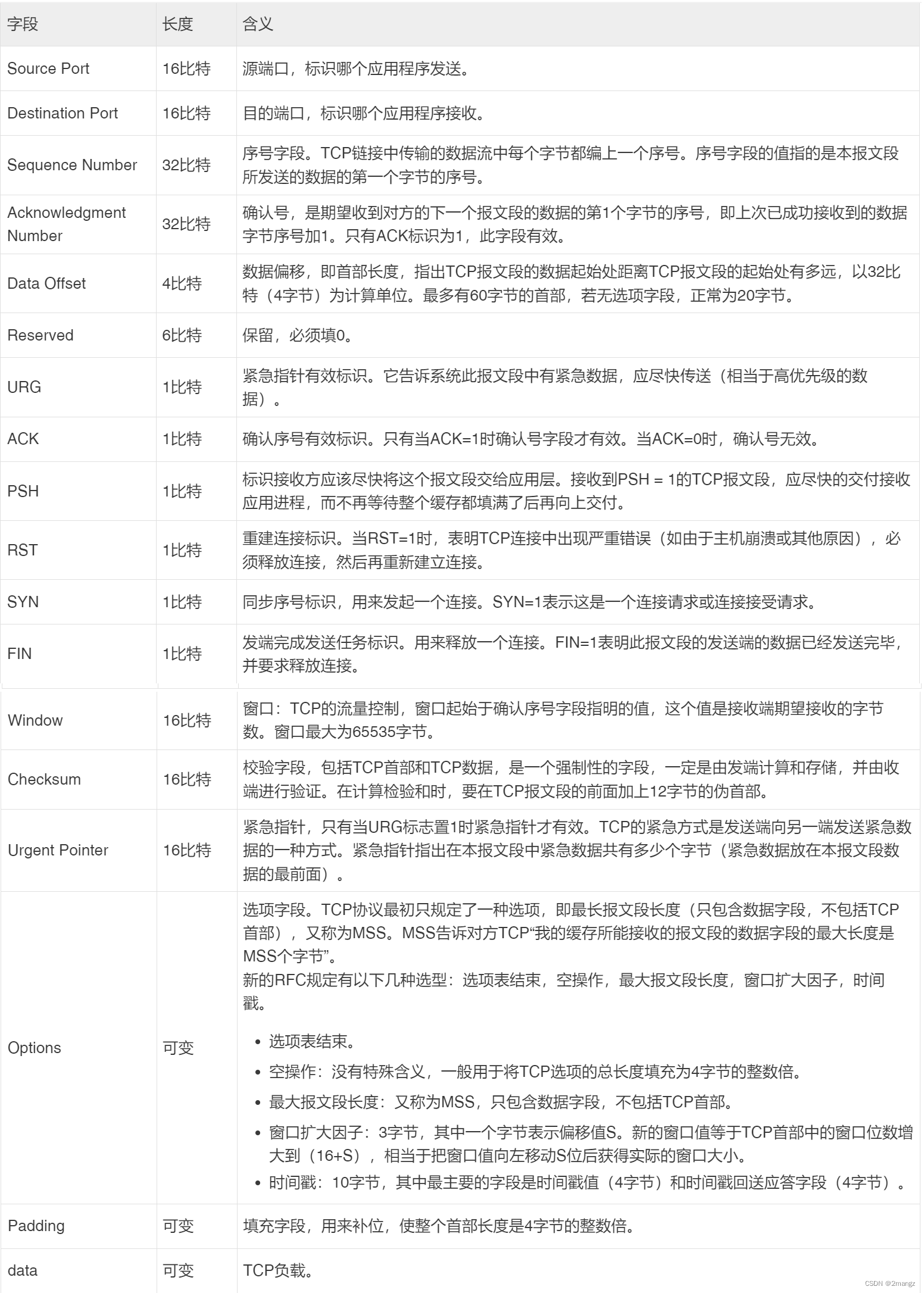
网络工程师必学知识:TCP抓包分析Seq、SYN、ACK的变化过程
网络工程师必学知识:TCP抓包分析Seq、SYN、ACK的变化过程 1.概述:2.抓包3.分析:重点1:重点2: 4.总结: 1.概述: 下面是我面试时遇到的问题。 问:TCP协议位于tcp/ip协议栈的哪一层呢? 答:这个问题要是答不上来,就不用看下面的内容了。 问:TCP抓包时Seq、SYN、ACK的变化过程? 答:见下面过程 下图是TCP Header Format: E

Word域代码学习(简单使用)-【SEQ】
Word域代码学习(简单使用)-【SEQ】 快捷键 序号快捷键操作1 Ctrl + F9 插入域代码花括号2 F9 显示域代码结果3 Shift + F9 切换为域代码4 Windows + Alt + F9 切换全部域代码 域代码说明 域代码不区分大小写在word中,依次选择插入➡文档部件➡域即可选择插入域代码 可以看到里面显示了许多种域类别 序列域

易基因:RNA-seq联合ChIP-seq分析揭示肝脏p53依赖的组织特异性抗辐射机制|抗性研究
大家好,这里是专注表观组学十余年,领跑多组学科研服务的易基因。 以前的研究发现胸腺和脾脏是体内辐射敏感组织,而肝脏不是。胸腺和脾脏在体内全身辐射后会触发p53依赖性凋亡,但这种肝脏特异性抗性的分子机制尚不清楚。 2023年03月28日,美国西奈山伊坎医学院James J. Manfredi团队通过对经辐射处理的小鼠器官进行联合RNA测序(RNA-seq)和染色质免疫沉淀测序(ChIP-seq)

单细胞RNA测序(scRNA-seq)Cellranger流程入门和数据质控
单细胞RNA测序(scRNA-seq)Cellranger流程入门和数据质控 单细胞RNA测序(scRNA-seq)基础知识可查看以下文章: 单细胞RNA测序(scRNA-seq)工作流程入门 单细胞RNA测序(scRNA-seq)细胞分离与扩增 1. 单细胞RNA-seq样本数据说明 样本数据来源文章:Acquired cancer resistance to combination
项目文章| Plant CellDAP-seq解析草莓NAC转录因子FvRIF的调控网络
DAP-seq是一种体外研究蛋白与DNA结合的技术,该技术利用麦胚乳表达体系表达目标蛋白然后与基因组DNA文库体外孵育,得到目标蛋白的结合信息。与ChIP-seq和CUT&Tag不同,DAP-seq不需要抗体,在植物中应用更为广泛。今天我们分享一篇2023年发表在The Plant Cell(影响因子:10.676)的文章“Deciphering the regulatory network

scFed:联邦学习用于scRNA-seq分类
scRNA-seq的出现彻底改变了我们对生物组织中细胞异质性和复杂性的理解。然而,大型,稀疏的scRNA-seq数据集的隐私法规对细胞分类提出了挑战。联邦学习提供了一种解决方案,允许高效和私有的数据使用。scFed是一个统一的联邦学习框架,允许在不侵犯数据隐私的情况下对四种分类算法进行基准测试,包括单细胞特定分类器和通用分类器。作者使用8个公开的具有不同大小、物种和技术的scRNA-seq数据集来
RNA-seq分析(Fastqc+Trimmomatic+STAR+HTseq-count+DESeq2)
最近做RNA-seq,正好把流程整理下,也希望分享和相互学习。 具体将以Fastqc + Trimmomatic + STAR + HTseq-count + DEseq2的流程来进行。 查看数据完整性 for dir in `ls`; do cd $dir; md5sum -c MD5_*txt; cd ..; done 预处理 FastQC + Trimmomatic fas
项目文章|IF=9.2 ChIP-seq解析转录因子STAT3在代谢性脂肪性肝病中的调控机制
华中科技大学同济医学院同济医院内科心内科吕家高和老年病科张存泰团队在《International Journal of Biological Sciences》(IF=9.2)杂志发表题为“The role of STAT3/VAV3 in glucolipid metabolism during the development of HFD-induced MAFLD”文章,通过ChIP