本文主要是介绍单细胞分析(Signac): PBMC scATAC-seq 整合,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
引言
在本教学指南中,我们将探讨由10x Genomics公司提供的人类外周血单核细胞(PBMCs)的单细胞ATAC-seq数据集。
加载包
首先加载 Signac、Seurat 和我们将用于分析人类数据的其他一些包。
if (!requireNamespace("EnsDb.Hsapiens.v75", quietly = TRUE))
BiocManager::install("EnsDb.Hsapiens.v75")
library(Signac)
library(Seurat)
library(EnsDb.Hsapiens.v75)
library(ggplot2)
library(patchwork)
与 scRNA-seq 数据整合
为了更好地理解 scATAC-seq 测序数据,我们可以通过与人类外周血单核细胞(PBMC)相关的单细胞 RNA 测序(scRNA-seq)实验来对细胞进行分类。我们采用了一种跨数据类型整合和标签迁移的方法,这种方法的详细教程可以在这里找到。我们的目标是找出基因活动矩阵与 scRNA-seq 数据集之间的共同相关性模式,以此来确定两种技术中对应的生物学状态。通过这种方法,我们可以为每个细胞在 scRNA-seq 定义的每个聚类标签下得到一个分类评分。
在本例中,我们加载了一个由 10x Genomics 提供的经过预处理的人类 PBMC 的 scRNA-seq 数据集。你可以从 10x Genomics 的官方网站下载这项实验的原始数据,也可以在 GitHub 上查看构建该数据对象所使用的代码。此外,你还可以直接下载已经预处理过的 Seurat 数据对象。
# Load the pre-processed scRNA-seq data for PBMCs
pbmc_rna <- readRDS("../vignette_data/pbmc_10k_v3.rds")
pbmc_rna <- UpdateSeuratObject(pbmc_rna)
transfer.anchors <- FindTransferAnchors(
reference = pbmc_rna,
query = pbmc,
reduction = 'cca'
)
## Running CCA
## Merging objects
## Finding neighborhoods
## Finding anchors
## Found 17505 anchors
predicted.labels <- TransferData(
anchorset = transfer.anchors,
refdata = pbmc_rna$celltype,
weight.reduction = pbmc[['lsi']],
dims = 2:30
)
## Finding integration vectors
## Finding integration vector weights
## Predicting cell labels
pbmc <- AddMetaData(object = pbmc, metadata = predicted.labels)
plot1 <- DimPlot(
object = pbmc_rna,
group.by = 'celltype',
label = TRUE,
repel = TRUE) + NoLegend() + ggtitle('scRNA-seq')
plot2 <- DimPlot(
object = pbmc,
group.by = 'predicted.id',
label = TRUE,
repel = TRUE) + NoLegend() + ggtitle('scATAC-seq')
plot1 + plot2
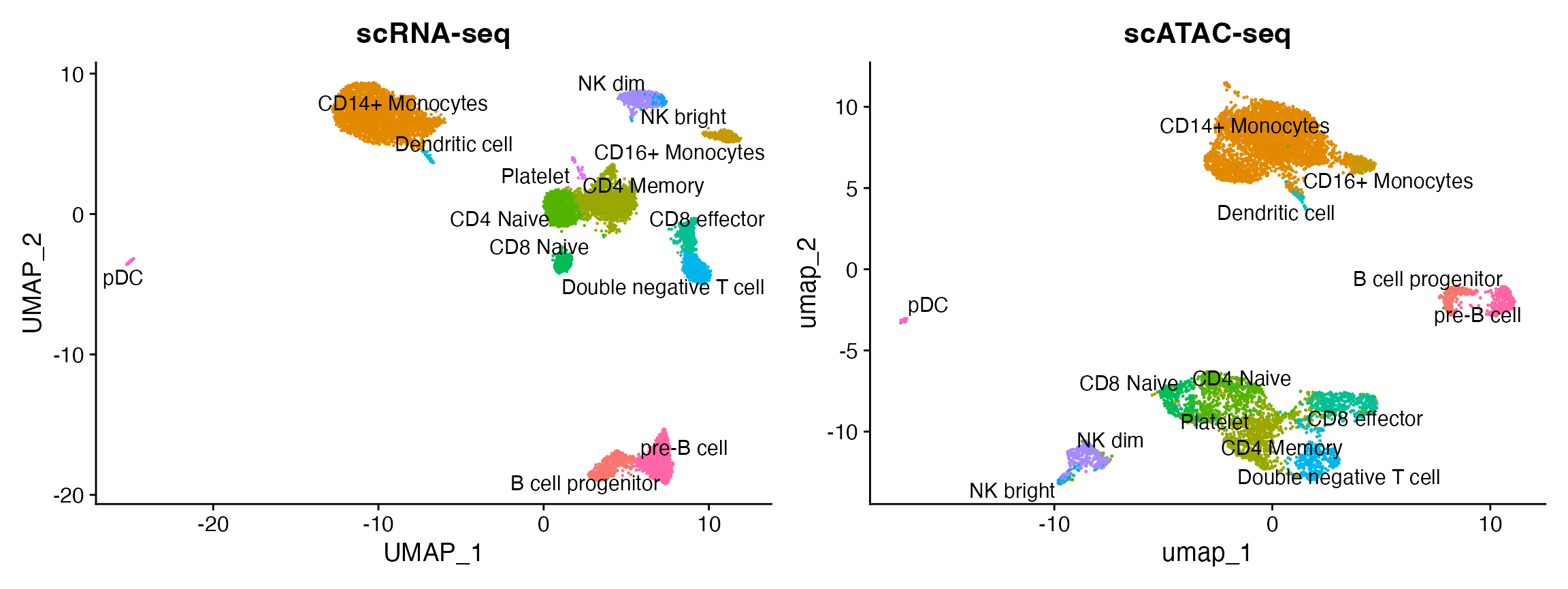
观察到基于单细胞 RNA 测序(scRNA)的分类结果与利用单细胞染色质可及性测序(scATAC-seq)数据生成的 UMAP(均匀流形近似和投影)图呈现出一致性。我们注意到,在 scATAC-seq 数据集中并未预测出血小板细胞,这是符合预期的,因为血小板是无核细胞,无法通过 scATAC-seq 技术被检测到。
为了整合我们的 scATAC-seq 聚类分析和标签转移结果,我们可以选择将每个聚类的名称重新指定为该聚类中最常见的预测标签。另外,我们也可以选择直接使用每个细胞的预测标签来进行后续分析。
# replace each label with its most likely prediction
for(i in levels(pbmc)) {
cells_to_reid <- WhichCells(pbmc, idents = i)
newid <- names(which.max(table(pbmc$predicted.id[cells_to_reid])))
Idents(pbmc, cells = cells_to_reid) <- newid
}
查找细胞类型之间差异可及的峰
为了识别不同细胞聚类间的差异开放区域,我们可以进行差异可及性(DA)分析。一种简便的方法是应用 Wilcoxon 秩和检验,Presto 软件包已经内嵌了一个非常高效的 Wilcoxon 检验程序,适用于 Seurat 数据对象的分析。
我们还可以使用逻辑回归进行差异性分析,这是 Ntranos 等人在 2018 年针对单细胞 RNA 测序(scRNA-seq)数据提出的建议,同时将片段总数作为一个隐变量来考虑,以减少测序深度差异对分析结果的影响。本研究将重点比较未激活的 CD4+ T 细胞和 CD14+ 单核细胞,但这些方法也适用于任何细胞群体的比较。此外,我们还可以使用 Seurat 提供的小提琴图、特征图、散点图、热图或其他任何可视化工具来展示这些特征峰。
# change back to working with peaks instead of gene activities
DefaultAssay(pbmc) <- 'peaks'
da_peaks <- FindMarkers(
object = pbmc,
ident.1 = "CD4 Naive",
ident.2 = "CD14+ Monocytes",
test.use = 'LR',
latent.vars = 'nCount_peaks'
)
head(da_peaks)
## p_val avg_log2FC pct.1 pct.2 p_val_adj
## chr14-99721608-99741934 1.414340e-280 5.571161 0.868 0.022 1.238410e-275
## chr14-99695477-99720910 5.529507e-222 5.100310 0.797 0.021 4.841691e-217
## chr17-80084198-80086094 8.962660e-221 7.032939 0.668 0.005 7.847795e-216
## chr7-142501666-142511108 6.506936e-212 4.757277 0.754 0.029 5.697538e-207
## chr2-113581628-113594911 5.746054e-188 -5.051770 0.035 0.663 5.031302e-183
## chr6-44025105-44028184 2.328105e-179 -4.394199 0.046 0.616 2.038512e-174
plot1 <- VlnPlot(
object = pbmc,
features = rownames(da_peaks)[1],
pt.size = 0.1,
idents = c("CD4 Naive","CD14+ Monocytes")
)
plot2 <- FeaturePlot(
object = pbmc,
features = rownames(da_peaks)[1],
pt.size = 0.1
)
plot1 | plot2
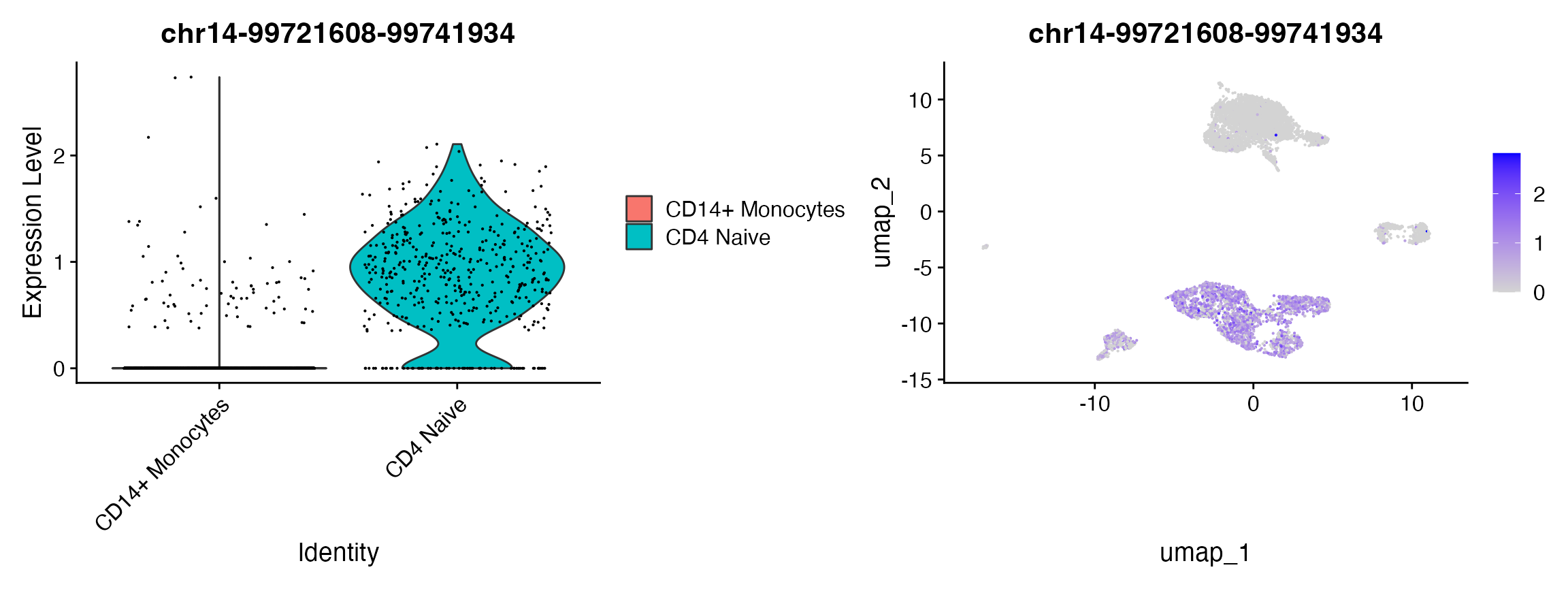
要识别两个细胞群体间的差异可访问区域,我们还可以通过比较这两组细胞的可访问性变化倍数来进行。这种方法比执行更复杂的差异可访问性测试要快捷,但它无法考虑到如细胞间总测序深度差异等潜在因素,并且不进行任何统计检验。尽管如此,这种方法仍然是一个有用的数据快速探索手段,可以通过 Seurat 软件包中的 FoldChange() 函数来实现。
fc <- FoldChange(pbmc, ident.1 = "CD4 Naive", ident.2 = "CD14+ Monocytes")
# order by fold change
fc <- fc[order(fc$avg_log2FC, decreasing = TRUE), ]
head(fc)
## avg_log2FC pct.1 pct.2
## chr6-28416849-28417227 11.45411 0.067 0
## chr7-110665002-110665493 11.41080 0.073 0
## chr8-19317420-19317942 11.22146 0.061 0
## chr1-172836553-172836955 11.20312 0.058 0
## chr2-191380525-191380926 11.15811 0.056 0
## chr8-90255778-90256179 11.03921 0.050 0
仅仅依靠峰值坐标可能难以进行分析解读。我们可以通过 ClosestFeature() 函数来识别每个峰值所对应的最近基因。在对基因列表进行深入分析时,可以发现,在未激活的 T 细胞(Naive T cells)中开放的峰值通常与如 BCL11B 和 GATA3(T 细胞分化的关键调控基因)等基因相邻,而在单核细胞(monocytes)中开放的峰值则通常与如 CEBPB(单核细胞分化的关键调控基因)等基因相邻。为了进一步深入分析,我们可以对 ClosestFeature() 函数返回的基因集进行基因本体(Gene Ontology, GO)富集分析,而目前有许多 R 语言包能够执行此类分析(例如,可以参考 GOstats 包)。
open_cd4naive <- rownames(da_peaks[da_peaks$avg_log2FC > 3, ])
open_cd14mono <- rownames(da_peaks[da_peaks$avg_log2FC < -3, ])
closest_genes_cd4naive <- ClosestFeature(pbmc, regions = open_cd4naive)
closest_genes_cd14mono <- ClosestFeature(pbmc, regions = open_cd14mono)
head(closest_genes_cd4naive)
## tx_id gene_name gene_id gene_biotype type
## ENST00000443726 ENST00000443726 BCL11B ENSG00000127152 protein_coding cds
## ENST00000357195 ENST00000357195 BCL11B ENSG00000127152 protein_coding cds
## ENST00000583593 ENST00000583593 CCDC57 ENSG00000176155 protein_coding cds
## ENSE00002456092 ENST00000463701 PRSS1 ENSG00000204983 protein_coding exon
## ENST00000546420 ENST00000546420 CCDC64 ENSG00000135127 protein_coding cds
## ENST00000455990 ENST00000455990 HOOK1 ENSG00000134709 protein_coding cds
## closest_region query_region distance
## ENST00000443726 chr14-99737498-99737555 chr14-99721608-99741934 0
## ENST00000357195 chr14-99697682-99697894 chr14-99695477-99720910 0
## ENST00000583593 chr17-80085568-80085694 chr17-80084198-80086094 0
## ENSE00002456092 chr7-142460719-142460923 chr7-142501666-142511108 40742
## ENST00000546420 chr12-120427684-120428101 chr12-120426014-120428613 0
## ENST00000455990 chr1-60280790-60280852 chr1-60279767-60281364 0
head(closest_genes_cd14mono)
## tx_id gene_name gene_id gene_biotype
## ENST00000432018 ENST00000432018 IL1B ENSG00000125538 protein_coding
## ENSE00001638912 ENST00000455005 RP5-1120P11.3 ENSG00000231881 lincRNA
## ENST00000445003 ENST00000445003 RP11-290F20.3 ENSG00000224397 lincRNA
## ENST00000568649 ENST00000568649 PPCDC ENSG00000138621 protein_coding
## ENST00000409245 ENST00000409245 TTC7A ENSG00000068724 protein_coding
## ENST00000484822 ENST00000484822 RXRA ENSG00000186350 protein_coding
## type closest_region query_region distance
## ENST00000432018 cds chr2-113593760-113593806 chr2-113581628-113594911 0
## ENSE00001638912 exon chr6-44041650-44042535 chr6-44025105-44028184 13465
## ENST00000445003 gap chr20-48884201-48894027 chr20-48889794-48893313 0
## ENST00000568649 cds chr15-75335782-75335877 chr15-75334903-75336779 0
## ENST00000409245 cds chr2-47300841-47301062 chr2-47297968-47301173 0
## ENST00000484822 gap chr9-137211331-137293477 chr9-137263243-137268534 0
本文由 mdnice 多平台发布
这篇关于单细胞分析(Signac): PBMC scATAC-seq 整合的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!







