本文主要是介绍inferCNV:scRNA-seq数据推断染色体拷贝数变化,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
inferCNV分析简介
inferCNV用于探索肿瘤单细胞RNA-Seq 数据,以确定体细胞大规模染色体拷贝数改变的证据,例如整个染色体或大片段染色体的增益或缺失。这是通过与一组参考“正常”细胞(这里的正常细胞可自行定义)进行比较,探索肿瘤基因组各部位基因的表达强度来完成的。热图展示每个染色体的相对表达强度,并且与“正常”细胞相比,肿瘤基因组的哪些区域过表达或降低(异常)。
同时我们需要了解两个概念:拷贝数变异(Copy number variation, CNV) 和 拷贝数改变(copy number alterations, CNA) 。
拷贝数改变(copy number alterations, CNA) :指的是基因组中的 DNA 片段在不同个体间的拷贝数存在变异。这些变异可以是片段的缺失(deletion)或扩增(duplication),并且这些片段通常较大,范围从 1 kb 到数 Mb 不等。
拷贝数改变(copy number alterations, CNA) :是指基因组中某些 DNA 片段的拷贝数发生变化,包括拷贝数的增加(扩增,amplification)和减少(缺失,deletion)。这些改变可以影响单个基因、多个基因甚至整个染色体区域, CNA 与CNV概念相似。
inferCNV分析的意义
我们对单细胞分析的时候会根据关键基因/标记进行细胞分群,基本上可以把大部分的细胞亚群给区分开来,但如果某些基因/标记广泛地存在于多种细胞亚群时,此时应进一步寻找其他的关键基因/标记进行区分,这种分析策略是“常规流程”。对于肿瘤细胞和正常细胞而言,我们常会遇到某些关键基因/标记在两者中均表达的情况(转录组水平),此时通过“常规流程”仍难以区分细胞的良恶性,因此换个角度来辅助区分良恶性细胞就显得尤为重要。
肿瘤细胞通常具有更多的CNV,而正常细胞则较为稳定,因此如果有一种工具能够可视化拷贝数变异的情况那么是不是就可以辅助鉴别良恶性细胞呢?基于这种情况inferCNV就应运而生了。
inferCNV分析结果详解
1、官方示例展示

图中的红色部分是References(Cells),也就是我们自行定义的对照(正常)细胞。这些对照细胞可以是相对于肿瘤细胞的正常细胞(癌与非癌),也可选用免疫细胞进行对照。
蓝色部分是Observations(Cells),也就是我们想要重点分析的细胞。
灰色部分是细胞的图注,上边的色块代表了对照细胞的情况,该图研究者纳入了Microglia/Macrophage和Oligodendrocytes(non-malignant)作为对照细胞,恶性细胞作为观察细胞。

红色方框部分代表的是分层聚类树,其中第一列色柱代表的是所有观察组细胞的分层聚类,第二列色柱则是与所提供输入的观察组细胞分类相对应。

第一列色柱根据分析时group_by_cluster参数设置的不同(True/False)会对结果产生差异:
如果group_by_cluster = FALSE ,意味着不按照研究者命名的cluster去分,换句话说就是第一列色柱是按照聚类树所切割成k_obs_groups分组的情况来表示树状图的细分。
如果group_by_cluster = TRUE ,左边的树状图是所有观察细胞的“线性连接”(树状图的根与每种类型的树状图的根相连,这导致它们都处于同一水平)。第一列色柱列是单一颜色,因为注释聚类时不使用k_obs_groups分组。
但实际函数中参数名称改成了cluster_by_groups,并且True/False的最终结果也对调过来了,这个可能跟R包更新了有关系,因此我个人意见是,只要看到第一条色柱是同一的颜色时,我们应当理解为不采用k_obs_groups分组,而当显示不同颜色时是采用了k_obs_groups分组。两种参数设置均可。
蓝色方框部分代表的是染色体的分组分别是从chr1-chr22,性染色体和线粒体染色体已经被过滤了。
红色和蓝色箭头分别代表染色体区域扩增和染色体区域缺失(两者均为异常)。

按照箭头看向对照和观察组细胞,在红色箭头处代表了malignant_MGH36细胞群,这群细胞相比于观察组(蓝色箭头) 在chr1, 4, 19中存在更显著的染色体缺失,在chr11, 21中存在更显著的染色体扩增。同样的我们可以看其他的三群恶性细胞,也均存在不同区域的染色体拷贝数异常。
2、其他数据展示—正常/肿瘤样本
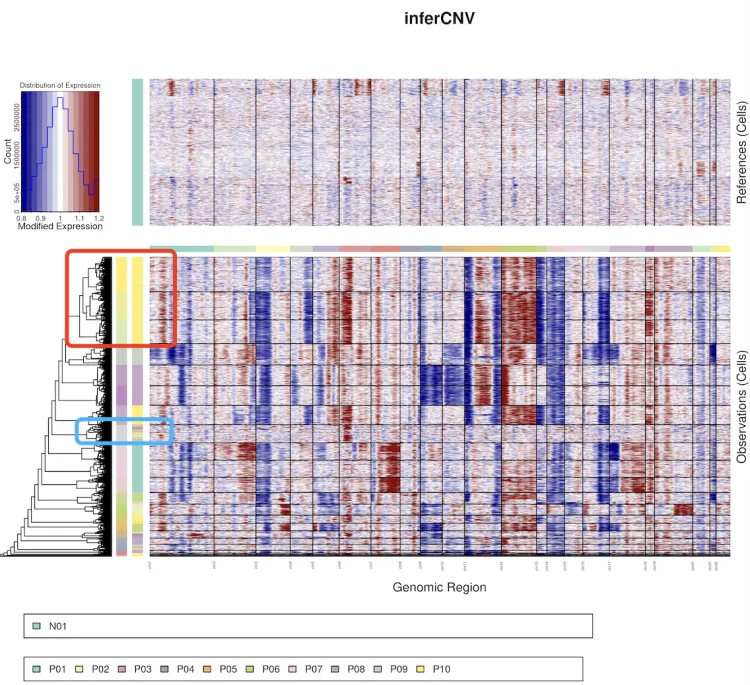
该结果显示1个正常样本作为对照组,10个肿瘤样本作为观察组。整体情况观察下来可以发现肿瘤组样本相比于对照组来说还是存在了很明显的染色体拷贝数异常。
红色方框中的第二列色柱是代表了P10肿瘤样本的信息,中间还混了一小部分的P05患者的信息,从聚类树分层后的色柱来看,P10肿瘤样本还可以进一步的细分为三群。
蓝色方框中的第二列色柱带了很多肿瘤样本的信息,但从聚类树分层后的色柱来看,这些肿瘤样本信息应当分成同一群。

同样的数据,修改了cluster_by_groups的值之后聚类树的图形出现了变化。在不按照k_obs_groups分组之后,主要根据每个样本内部的情况进行分组,我们可以明显发现不同样本内部也存在着很大的异质性(红色方框)。
3、其他数据展示—细胞亚群

该结果显示前期分析得到了17个细胞亚群,其中第15个亚群根据关键基因/标记推测认为可能是正常细胞群,因此通过inferCNV进行辅助分析判断第15个亚群是否是正常细胞群,从结果来看确实也符合正常细胞亚群的猜测。
而根据聚类树结果来看,k_obs_groups分组和细胞亚群分组一致。
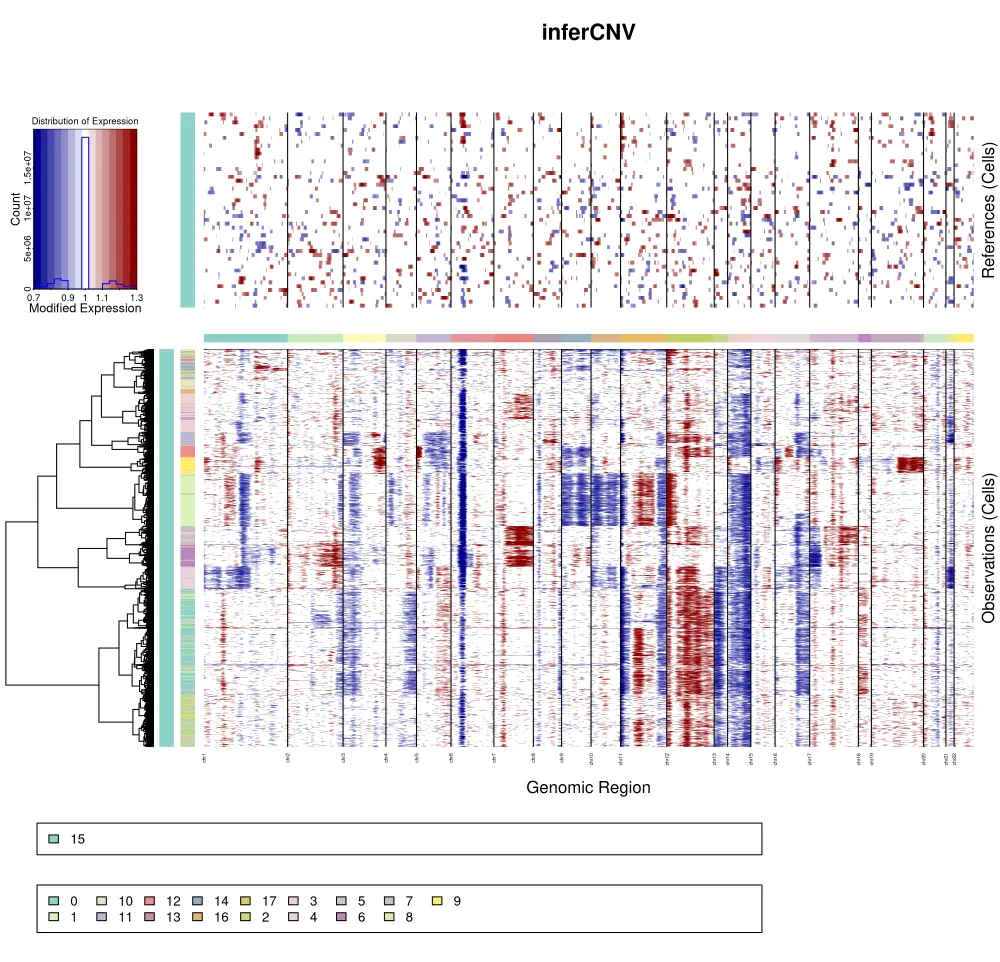
在不按照k_obs_groups分组之后,我们可以明显发现很多细胞亚群之间没有十分明显的界限,也就是说这个结果提示我们在后续聚类的时候可以把很多细胞亚群合并成同一群。
inferCNV分析流程
1.加载对象和R包
rm(list = ls())
library(infercnv)
library(Seurat)
library(gplots)
library(ggplot2)
#这里加载的是seurat对象,替换自己的数据即可
load("sce_CNV_N_T.Rdata") #检查一下自己导入进来的数据
DimPlot(sce,reduction = 'umap',label = TRUE,pt.size = 0.5) +NoLegend()
2.读取数据
#根据需求去检查一下数据集的信息
table(Idents(sce))
table(sce@meta.data$seurat_clusters)
table(sce@meta.data$orig.ident)
#table(sce@meta.data$patient)
#table(sce@meta.data$ID)# 文件制作1:表达量矩阵
# 除了用GetAssayData函数其实也可以直接sce@assays$RNA@count即可
dat <- GetAssayData(sce,layer = 'counts',assay = 'RNA')
dat[1:4,1:4]# 文件制作2:样本的描述
groupinfo <- data.frame(v1 = colnames(dat),v2 = sce@meta.data$patient)# 文件制作3:基因在染色体中的坐标
library(AnnoProbe)
#annoGene 函数返回一个数据框,包含输入基因的详细注释信息。
#注释信息可能包括基因名称、染色体位置、基因描述等。
geneInfor <- annoGene(rownames(dat),"SYMBOL","human") #物种需要小心哦
# 使用逻辑条件来移除包含 chrM, chrX, 和 chrY 的行
geneInfor <- geneInfor[!geneInfor$chr %in% c("chrM", "chrX", "chrY"), ]
#提取chr后后面的数字并转化为num,从而按这个num排序
#sub函数用于把chr替换为空
geneInfor$chr_num <- as.numeric(sub("chr", "", geneInfor$chr))
colnames(geneInfor)#with函数的作用是简化写法,可问gpt
geneInfor <- geneInfor[with(geneInfor,order(chr_num,start)),c(1,4:6)]
geneInfor <- geneInfor[!duplicated(geneInfor[,1]),]length(unique(geneInfor[,1]))
head(geneInfor)
3.排序一下
#保留行名在geneInfor第一列中存在的行。
dat <- dat[rownames(dat) %in% geneInfor[,1],]
#match(x, table):match函数返回x中每个元素在table中的位置索引。
#获得位置后对dat进行重新排序使其跟geneInfor中的顺序一致
dat <- dat[match(geneInfor[,1],rownames(dat)),]
#检查信息
dim(dat)
head(groupinfo)
dat[1:4,1:4]
table(groupinfo$v2)
dim(groupinfo)
4.保存/输出文件
#统一把”-“改成”_“,因为后续运行inferCNV的时候需要读取保存文档,若不修改则会出现错误。
#expFile:是一个变量,存储写入的文件的文件名或路径。在这里文件名是expFile.txt。
expFile <- 'expFile.txt' #定义输出文件名
colnames(dat) <- gsub("-", "_", colnames(dat))
write.table(dat, file = expFile, sep = '\t', quote = F)#groupFiles:是一个变量,存储写入的文件的文件名或路径。在这里文件名是groupFiles.txt。
groupFiles <- 'groupFiles.txt'
groupinfo$v1 <- gsub("-", "_", groupinfo$v1)
write.table(groupinfo,file = groupFiles, sep = '\t',quote = F, col.names = F, row.names = F)#geneFile:是一个变量,存储写入的文件的文件名或路径。在这里文件名是geneFile.txt。
head(geneInfor)
geneFile <- 'geneFile.txt'
write.table(geneInfor, file = geneFile, sep = '\t',quote = F, col.names = F, row.names = F)
5. inferCNV分析
#创建对象,请注意文件需要一一对应哦!
#ref_group_names 这里的细胞是正常对照,然后跟其他的细胞比较
infercnv_obj <- CreateInfercnvObject(raw_counts_matrix = expFile,annotations_file = groupFiles,delim = "\t",gene_order_file = geneFile,ref_group_names = c("N01")
)infercnv_obj2 <- infercnv::run(infercnv_obj,cutoff = 0.1, #smart-seq选择1,10X选择0.1out_dir = "infercnv_output", # dir is auto# 是否根据细胞注释文件的分组# 对肿瘤细胞进行分组# 影响read.dendrogram, 如果有多个细胞类型,且设置为TRUE,# 后续的read.dendrogram无法执行cluster_by_groups = F, #是否根据患者类型(由细胞注释文件中定义)hclust_method = "ward.D2",# ward.D2 方法进行层次聚类analysis_mode = "samples", # 默认是samples,推荐是subclustersdenoise = TRUE, # 去噪音HMM = F, ##特别耗时间,是否要去背景噪音plot_steps = F, #不在每个步骤后生成图形。leiden_resolution = "auto", #可以手动调参数num_threads = 4 #4线程工作,加快速度)

接下来使用曾老师的方法去检查拷贝数变异的情况
6、读取数据加载R包
rm(list = ls())
options(stringsAsFactiors = F)
library(phylogram)
library(gridExtra)
library(grid)
require(dendextend)
require(ggthemes)
library(tidyverse)
library(Seurat)
library(infercnv)
library(miscTools)load("sce_CNV_N_T.Rdata")
7、把inferCNV中的run.final.infercnv_obje文件读取进来
infer_CNV_obj<-readRDS('../3.inferCNV_N_T/infercnv_output/run.final.infercnv_obj')
expr <- infer_CNV_obj@expr.data
expr[1:4,1:4]
data_cnv <- as.data.frame(expr)
dim(expr)
colnames(data_cnv)
rownames(data_cnv)#记得要改一下sce中的列名
colnames(sce) <- gsub("-","_",colnames(sce))
meta <- sce@meta.data
8、计算CNV并绘图
#要根据参数修改哦,我这里的对照组只有N01
if(T){tmp1 = expr[,infer_CNV_obj@reference_grouped_cell_indices$N01]tmp = tmp1#tmp2 = expr[,infer_CNV_obj@reference_grouped_cell_indices$`ref-2`]#tmp= cbind(tmp1,tmp2)down=mean(rowMeans(tmp)) - 2 * mean( apply(tmp, 1, sd))up=mean(rowMeans(tmp)) + 2 * mean( apply(tmp, 1, sd))oneCopy=up-downoneCopya1= down- 2*oneCopya2= down- 1*oneCopydown;upa3= up + 1*oneCopya4= up + 2*oneCopy cnv_score_table<-infer_CNV_obj@expr.datacnv_score_table[1:4,1:4]cnv_score_mat <- as.matrix(cnv_score_table)# Scoringcnv_score_table[cnv_score_mat > 0 & cnv_score_mat < a2] <- "A" #complete loss. 2ptscnv_score_table[cnv_score_mat >= a2 & cnv_score_mat < down] <- "B" #loss of one copy. 1ptscnv_score_table[cnv_score_mat >= down & cnv_score_mat < up ] <- "C" #Neutral. 0ptscnv_score_table[cnv_score_mat >= up & cnv_score_mat <= a3] <- "D" #addition of one copy. 1ptscnv_score_table[cnv_score_mat > a3 & cnv_score_mat <= a4 ] <- "E" #addition of two copies. 2ptscnv_score_table[cnv_score_mat > a4] <- "F" #addition of more than two copies. 2pts# Checktable(cnv_score_table[,1])# Replace with score cnv_score_table_pts <- cnv_score_matrm(cnv_score_mat)# cnv_score_table_pts[cnv_score_table == "A"] <- 2cnv_score_table_pts[cnv_score_table == "B"] <- 1cnv_score_table_pts[cnv_score_table == "C"] <- 0cnv_score_table_pts[cnv_score_table == "D"] <- 1cnv_score_table_pts[cnv_score_table == "E"] <- 2cnv_score_table_pts[cnv_score_table == "F"] <- 2cnv_score_table_pts[1:4,1:4]str(as.data.frame(cnv_score_table_pts[1:4,1:4])) cell_scores_CNV <- as.data.frame(colSums(cnv_score_table_pts))colnames(cell_scores_CNV) <- "cnv_score"
}#可视化
head(cell_scores_CNV)
score=cell_scores_CNV
head(score)
meta$totalCNV = score[match(colnames(sce),rownames(score)),1]
p <- ggplot(meta, aes(x= patient, y=totalCNV, fill=patient)) +geom_boxplot();print(p)
ggsave("totalCNV.png",plot = p, width = 8,height = 6,dpi = 300)

补充资料:
官方说明:
https://github.com/broadinstitute/inferCNV/wiki
技能树推文及B站视频:
推文名称:肿瘤单细胞转录组拷贝数分析结果解读和应用
https://www.bilibili.com/video/BV19Q4y1R7cu/?spm_id_from=333.999.0.0&vd_source=3a13860df939bc922ad1fd6099e42c1d
致谢:感谢曾老师,小洁老师以及生信技能树团队全体成员(部分代码来源:生信技能树马拉松和数据挖掘课程)。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(希望多多交流)。更多内容可关注公众号:生信方舟
- END -
这篇关于inferCNV:scRNA-seq数据推断染色体拷贝数变化的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!





