本文主要是介绍Signac 单细胞|ATAC-seq Call peak,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
引言
本文将向您展示如何利用MACS2软件,在单细胞ATAC-seq的基因组数据中识别基因调控区域的峰值。
实战
在使用Signac进行峰值检测之前,您需要先安装MACS2。您可以通过pip或conda安装它,或者从源代码自行编译。
本次演示以人类外周血单核细胞的单细胞ATAC-seq数据为例。首先,请加载必要的软件包和预先处理过的Seurat数据对象。
library(Signac)
library(Seurat)
pbmc <- readRDS("../vignette_data/pbmc.rds")
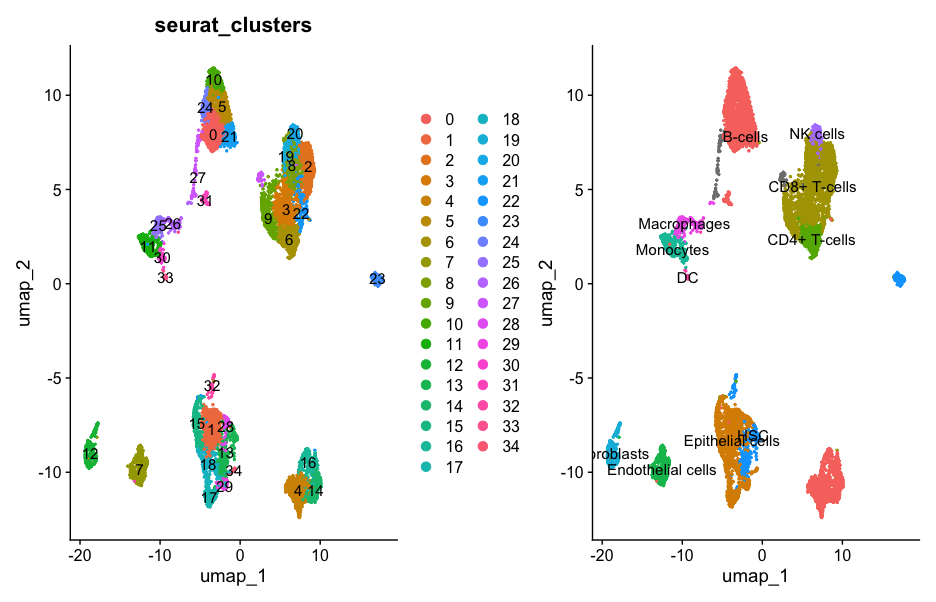
DimPlot(pbmc)

使用 CallPeaks() 函数可以进行峰值检测,这可以针对不同的细胞群体单独进行,或者综合所有细胞的数据来完成。若要在每个已标记的细胞类型上识别峰值,我们可以通过 group.by 参数来实现。
peaks <- CallPeaks(
object = pbmc,
group.by = "predicted.id"
)
结果以 GRanges 对象的形式返回,并带有一个附加元数据列,列出了每个峰在其中识别的细胞类型:

要计算每个峰值区域的计数,您可以利用 FeatureMatrix() 函数来实现。
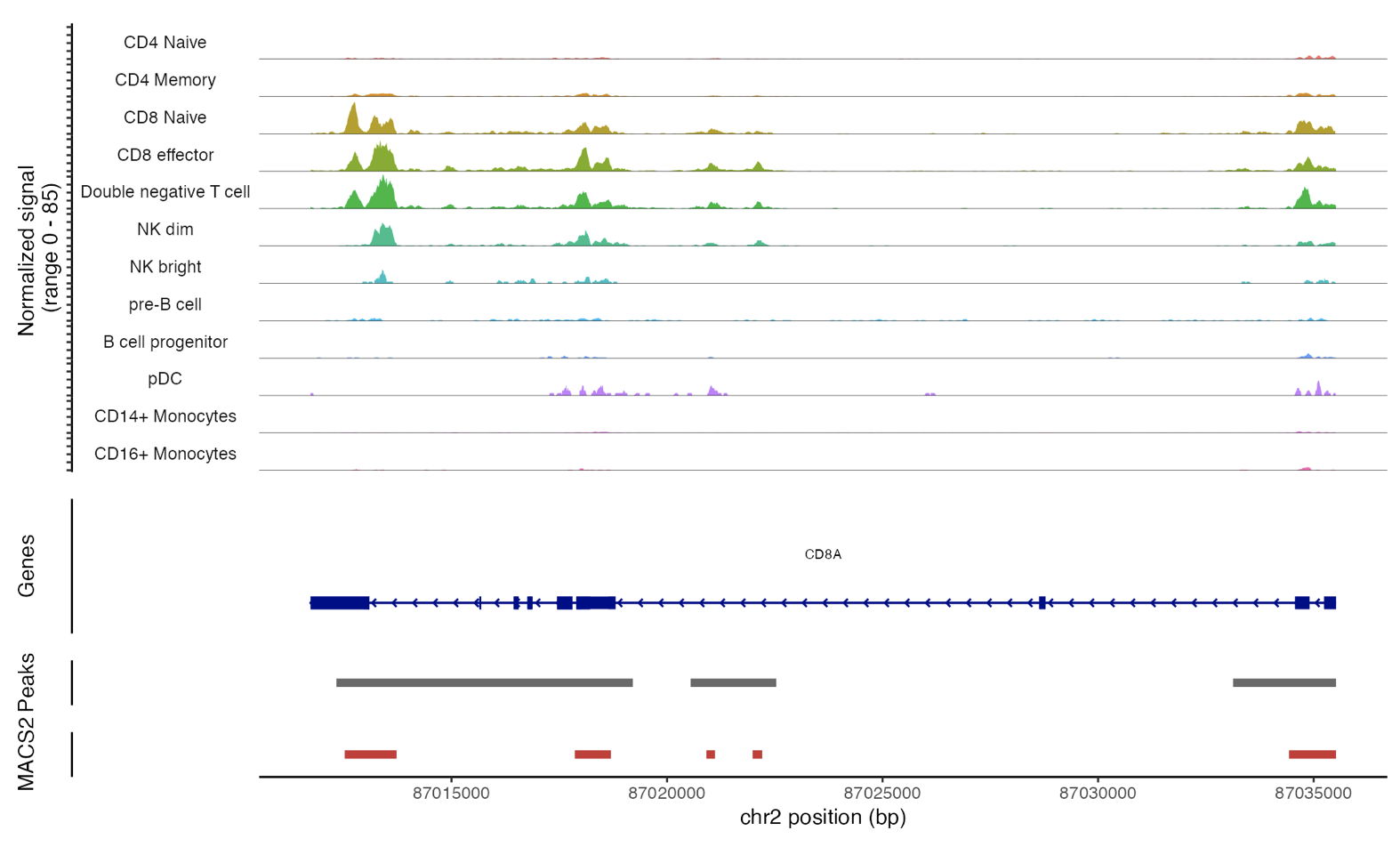
通过 CoveragePlot() 函数,我们可以将特定于细胞类型的 MACS2 峰值检测结果与 10x Cellranger 的峰值检测结果(目前 pbmc 对象中正在使用的)进行可视化对比。在图中,Cellranger 的峰值以灰色表示,而 MACS2 的峰值则以红色标注。
CoveragePlot(
object = pbmc,
region = "CD8A",
ranges = peaks,
ranges.title = "MACS2"
)
## Warning: Removed 1 row containing missing values or values outside the scale range
## (`geom_segment()`).
本文由 mdnice 多平台发布
这篇关于Signac 单细胞|ATAC-seq Call peak的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!