本文主要是介绍RNA-seq上下游分析snakemake流程,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
学习完snakemake后写的第一个流程是RNA-seq上游定量和下游的质控和差异分析。
使用fastp处理fastq文件,在使用START比对到基因组同时得到raw count,使用非冗余外显子长度作为基因的长度计算FPKM、TPM,同时也生成了CPM的结果。
非冗余外显子长度计算可以参考之前的推文转录组实战02: 计算非冗余外显子长度之和
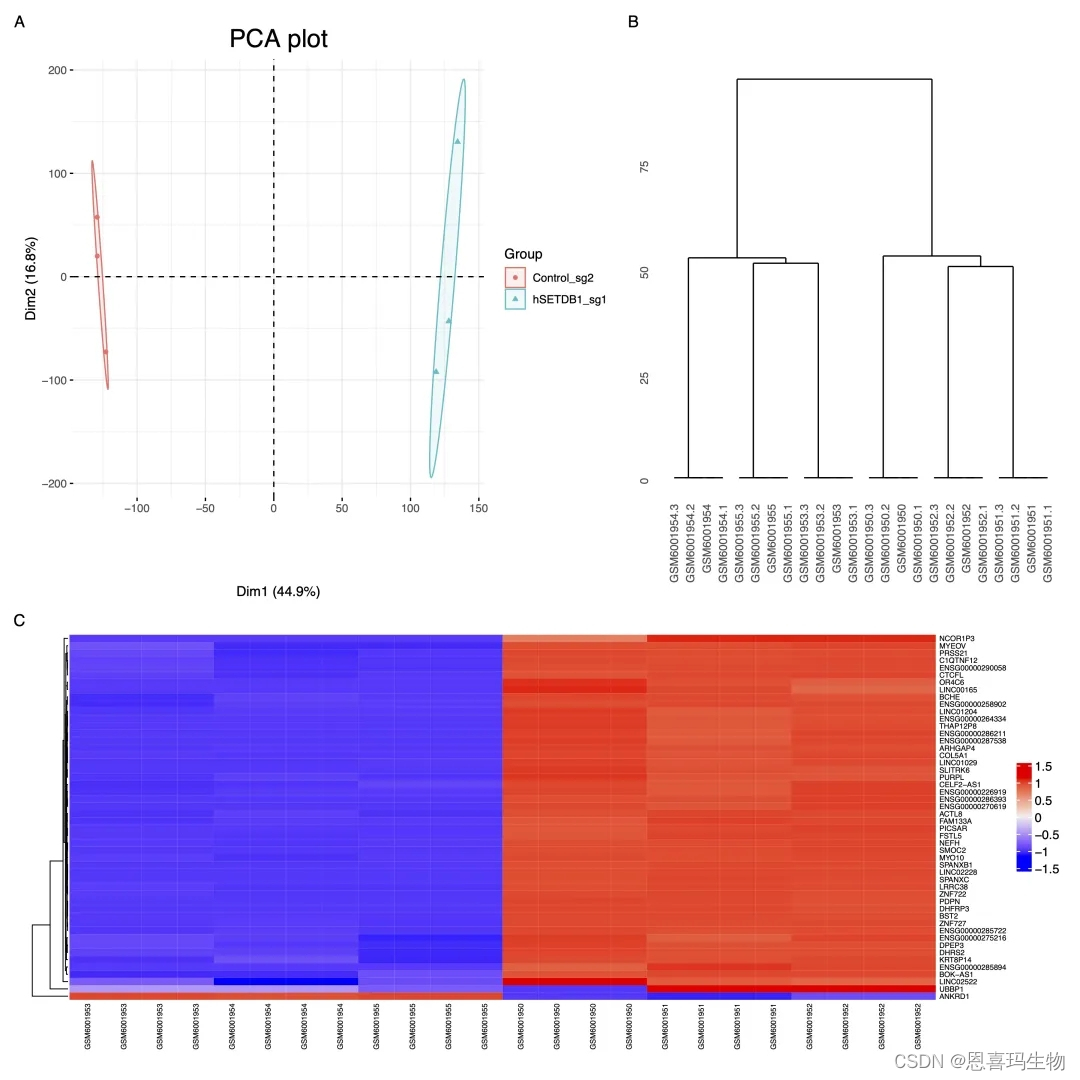
对定量结果质控使用生信技能树的三张图(PCA、树状图、热图)。
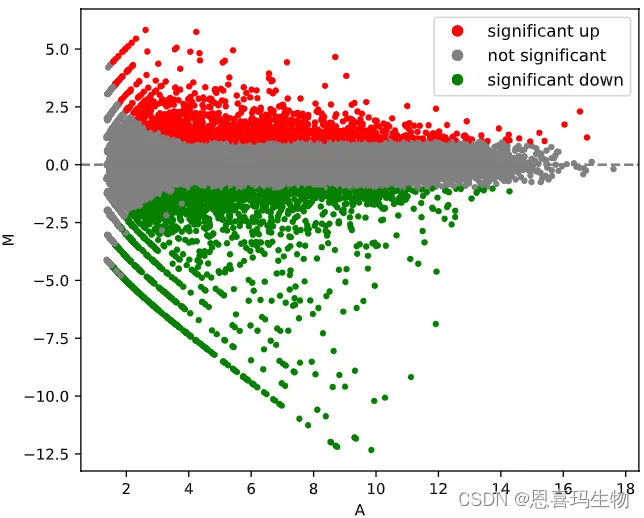
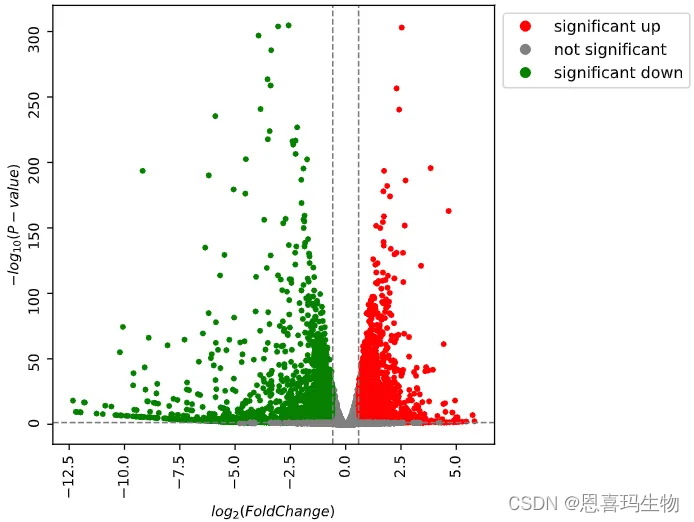
使用python版的DEseq2对组间做差异分析(火山图和MA图)。
流程代码在https://jihulab.com/BioQuest/SnakeMake-RNA-seq 或https://github.com/BioQuestX/SnakeMake-RNA-seq
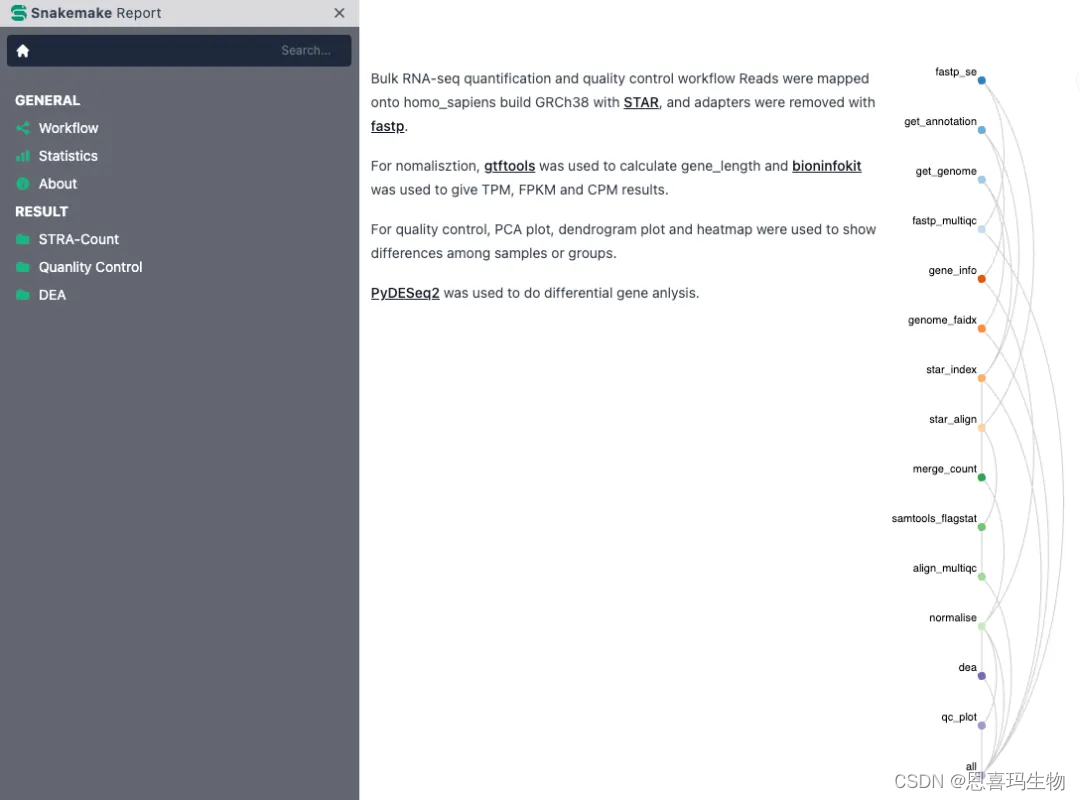
A SnakeMake workflow for Bulk RNA-seq
Reads were mapped onto ensembl genome with STAR, and adapters were removed with fastp.
For nomalisztion, gtftools was used to calculate gene_length and bioninfokit was used to give TPM, FPKM and CPM results.
For quality control, PCA plot, dendrogram plot and heatmap were used to show differences among samples or groups.
PyDESeq2 was used to perform differential expression anlysis.
General settings
To configure this workflow, modify config/config.yaml according to your needs, following the explanations provided in the file.
Sample sheet
-
Add samples to
config/samples.tsv. Only the columnSampleis mandatory, but any additional columns can be added. -
For each sample, add one or more sequencing units (runs, lanes or replicates) to the
Unitcolumn ofconfig/samples.tsv. -
For each sample, define
Groupcolumn(experimental or clinical attribute).
Report
这篇关于RNA-seq上下游分析snakemake流程的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!





