snakemake专题
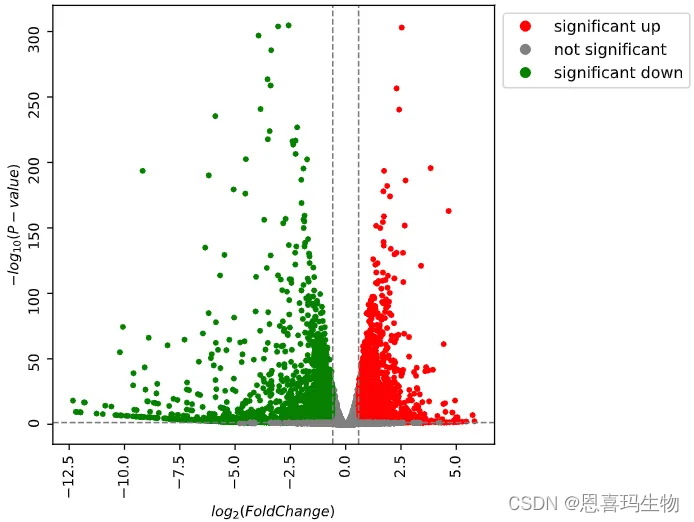
RNA-seq上下游分析snakemake流程
学习完snakemake后写的第一个流程是RNA-seq上游定量和下游的质控和差异分析。 使用fastp处理fastq文件,在使用START比对到基因组同时得到raw count,使用非冗余外显子长度作为基因的长度计算FPKM、TPM,同时也生成了CPM的结果。 非冗余外显子长度计算可以参考之前的推文转录组实战02: 计算非冗余外显子长度之和 对定量结果质控使用生信技能树的三张图(PCA、树

snakemake: 常用安装方法推荐,及详细安装步骤
Snakemake 的安装推荐使用 Conda 管理器,因为 Conda 可以方便地管理软件包及其依赖,并且可以创建隔离的环境以避免版本冲突。以下是使用 Conda 安装 Snakemake 的推荐步骤,包括如何首先安装 Conda(如果你还没有安装的话)。 安装 Miniconda 下载 Miniconda: 访问 Miniconda 的官网 并根据你的操作系统下载相应版本的 Minicon
SnakeMake介绍:主要特性、工作原理、应用场景
Snakemake 是一种基于 Python 的工作流管理工具,广泛用于生物信息学领域以及其他需要复杂数据分析的科学研究中。它结合了 Makefile 的特性,并引入了灵活的特性,使得自动化数据分析变得更加简单和高效。以下是关于 Snakemake 的详细介绍,包括其主要特性、工作原理和应用场景。 主要特性 易于学习的语法:Snakemake 使用类似于 Python 的语法,使得编写和理解工

Snakemake:初探
我已经安装了mamba(没有的话可以这样试试) curl -L https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-Linux-x86_64.sh -o Mambaforge-Linux-x86_64.shbash Mambaforge-Linux-x86_64.sh 安装snakemake
用snakemake进行RNAseq分析
1.下载sra文件 prefetch SRR* -O {outputfile}prefetch -O output --option-file SRR_Acc_List.txt 2.sra文件转换fa -- gzip 转换fa.gz #定义存放输出数据的文件夹,需要先创建这个文件夹‘fastq’mkdir fastqfqdir=/trainee2/Mar7/rna/pro