本文主要是介绍GATK4 somatic mutations analysis,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
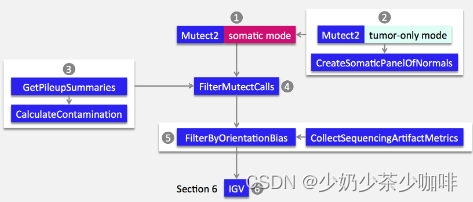
Section 1 用Mutect2调用体细胞突变
Section 2 概述如何使用Mutect2的纯肿瘤模式创建panel of normal resource
Section 3 概述了如何估计交叉样本污染
Section 4 显示了如何用FilterMutectCalls过滤callset。与GATK3不同,GATK4中,体细胞调用和过滤由不同的工具来体现
Section 5 显示了一个可选的过滤步骤,以过滤带有方向性偏差的序列背景artifacts
Section 6 显示了如何在IGV中设置手动观察
参考基因组resource:
GATK Resource Bundle
gs://gatk-best-practices/somatic-hg38
1. call somatic short variants and generate a bamout with Mutect2
这里我们有一个相当复杂的命令,使用Mutect2调用HCC1143肿瘤样本上的体细胞变异。关于体细胞调用的概要,见这篇文章。该命令调用了肿瘤样本中的体细胞变异,并使用了一个匹配的正常人、一个正常人面板(PoN)和一个群体生殖系变异资源。Here we have a rather complex command to call somatic variants on the HCC1143 tumor sample using Mutect2. For a synopsis of what somatic calling entails, see this article. The command calls somatic variants in the tumor sample and uses a matched normal, a panel of normals (PoN) and a population germline variant resource.
gatk --java-options "-Xmx2g" Mutect2 \-R hg38/Homo_sapiens_assembly38.fasta \-I tumor.bam \-I normal.bam \-tumor HCC1143_tumor \-normal HCC1143_normal \-pon resources/chr17_pon.vcf.gz \--germline-resource resources/chr17_af-only-gnomad_grch38.vcf.gz \--af-of-alleles-not-in-resource 0.0000025 \--disable-read-filter MateOnSameContigOrNoMappedMateReadFilter \-L chr17plus.interval_list \-O 1_somatic_m2.vcf.gz \-bamout 2_tumor_normal_m2.bam 这将产生一个未经过滤的体细胞呼叫集1_somatic_m2.vcf.gz,一个重新组装的读数BAM 2_tumor_normal_m2.bam和各自的索引1_somatic_m2.vcf.gz.tbi和2_tumor_normal_m2.bai。
Comments on select parameters
用两个参数指定体细胞调用的病例样本。用-I提供BAM,用-tumor提供样本的读组样本名称(SM字段值)。使用GetSampleName来查找读组SM字段。或者,使用Samtools view -H tumor.bam | grep '@RG'。Specify the case sample for somatic calling with two parameters. Provide the BAM with -I and the sample's read group sample name (the SM field value) with -tumor. To look up the read group SM field use GetSampleName. Alternatively, use samtools view -H tumor.bam | grep '@RG'.
预先过滤控制样本排列中的变异点。用-I指定对照BAM,用-normal指定对照样本的读组样本名称(SM字段值)。在肿瘤与正常对照相匹配的情况下,我们甚至可以排除罕见的种系变异和个体特异性的伪影。如果我们用Mutect2分析我们的肿瘤样本,而没有匹配的正常样本,我们得到的调用比匹配的正常样本多一个数量级。Prefilter variant sites in a control sample alignment. Specify the control BAM with -I and the control sample's read group sample name (the SM field value) with -normal. In the case of a tumor with a matched normal control, we can exclude even rare germline variants and individual-specific artifacts. If we analyze our tumor sample with Mutect2 without the matched normal, we get an order of magnitude more calls than with the matched normal.
预先过滤常模调用集中的变异点。用-pon指定常模面板(PoN)的VCF。第2节概述了如何创建一个PoN。常态面板不仅代表了常见的种系变异位点,它还展示了测序数据中常见的噪声位点,如测序中的映射神器或其他有些随机但系统的神器。默认情况下,该工具不会重新组合也不会发出与PoN变体完全匹配的变体位点。要启用PoN位点的基因分型,请使用--基因分型-on-sites选项。如果匹配不准确,例如有一个等位基因不匹配,该工具会重新组合该区域,发出呼叫并在INFO字段中用IN_PON注释匹配。Prefilter variant sites in a panel of normals callset. Specify the panel of normals (PoN) VCF with -pon. Section 2 outlines how to create a PoN. The panel of normals not only represents common germline variant sites, it presents commonly noisy sites in sequencing data, e.g. mapping artifacts or other somewhat random but systematic artifacts of sequencing. By default, the tool does not reassemble nor emit variant sites that match identically to a PoN variant. To enable genotyping of PoN sites, use the --genotype-pon-sites option. If the match is not exact, e.g. there is an allele-mismatch, the tool reassembles the region, emits the calls and annotates matches in the INFO field with IN_PON.
通过用--种系资源指定一个种群种系资源--germline-resource来注释变异等位基因。种系资源必须包含特定的等位基因频率,即它必须包含INFO字段中的AF注释[4]。该工具用群体等位基因频率来注释变异的等位基因。当使用群体种系资源时,可以考虑将--af-of-alleles-not-in-resource参数从其默认的0.001调整。例如,gnomAD资源af-only-gnomad_grch38.vcf.gz代表了~20万个外显子和~16万个基因组,而教程数据是外显子数据,所以我们将--af-of-alleles-not-in-resource调整为0.0000025,相当于1/(2*外显子样本)。默认的0.001适合于没有任何群体资源的人类样本分析。它是基于人类的平均杂合率。群体等位基因频率(POP_AF)和非资源性等位基因的因素在变异体的概率计算中。Annotate variant alleles by specifying a population germline resource with --germline-resource. The germline resource must contain allele-specific frequencies, i.e. it must contain the AF annotation in the INFO field [4]. The tool annotates variant alleles with the population allele frequencies. When using a population germline resource, consider adjusting the --af-of-alleles-not-in-resource parameter from its default of 0.001. For example, the gnomAD resource af-only-gnomad_grch38.vcf.gz represents ~200k exomes and ~16k genomes and the tutorial data is exome data, so we adjust --af-of-alleles-not-in-resource to 0.0000025 which corresponds to 1/(2*exome samples). The default of 0.001 is appropriate for human sample analyses without any population resource. It is based on the human average rate of heterozygosity. The population allele frequencies (POP_AF) and the af-of-alleles-not-in-resource factor in probability calculations of the variant being somatic.
包括那些配偶映射到不同Contig的读数。对于我们使用alt-aware和alt后处理的对准GRCh38的体细胞分析,我们用-disable-read-filter MateOnSameContigOrNoMappedMateReadFilter禁用一个特定的读数过滤器。这个过滤器会从分析中删除那些配偶映射到不同contig的配对读数。由于BWA对那些与交替的contig(对GRCh38的alt-aware映射)对齐较好的配偶信息进行纵横交错的方式,我们希望将这些类型的读数纳入我们的分析中。否则,我们可能会错过检测与交替单倍型相关的SNVs和indels。禁用这个过滤器偏离了目前的生产实践。Include reads whose mate maps to a different contig. For our somatic analysis that uses alt-aware and post-alt processed alignments to GRCh38, we disable a specific read filter with --disable-read-filter MateOnSameContigOrNoMappedMateReadFilter. This filter removes from analysis paired reads whose mate maps to a different contig. Because of the way BWA crisscrosses mate information for mates that align better to alternate contigs (alt-aware mapping to GRCh38), we want to include these types of reads in our analysis. Otherwise, we may miss out on detecting SNVs and indels associated with alternate haplotypes. Disabling this filter deviates from current production practices.
用-L参数将分析锁定在特定的基因组区间。在这里,我们指定这个选项来加快我们在小型教程数据上的运行速度。对于我们在第4节中使用的完整调用集,调用的是整个数据,没有区间文件。Target the analysis to specific genomic intervals with the -L parameter. Here we specify this option to speed up our run on the small tutorial data. For the full callset we use in section 4, calling was on the entirety of the data, without an intervals file.
用-bamout生成重新组装的排列组合文件。bamout排列包含了正常人和肿瘤的人工单倍型和重新组合的排列,可以手动审查调用。这个参数不是工具所要求的,但推荐使用,因为添加这个参数只需要花费总运行时间的一小部分。Generate the reassembled alignments file with -bamout. The bamout alignments contain the artificial haplotypes and reassembled alignments for the normal and tumor and enable manual review of calls. The parameter is not required by the tool but is recommended as adding it costs only a small fraction of the total run time.
为了说明Mutect2是如何应用注释的,下面是全部调用集中的五个多平行基因点。用gzcat somatic_m2.vcf.gz | awk '$5 ~", "拉出这些。awk '$5 ~","'对第5列中含有逗号的记录进行子集。To illustrate how Mutect2 applies annotations, below are five multiallelic sites from the full callset. Pull these out with gzcat somatic_m2.vcf.gz | awk '$5 ~","'. The awk '$5 ~","' subsets records that contain a comma in the 5th column.
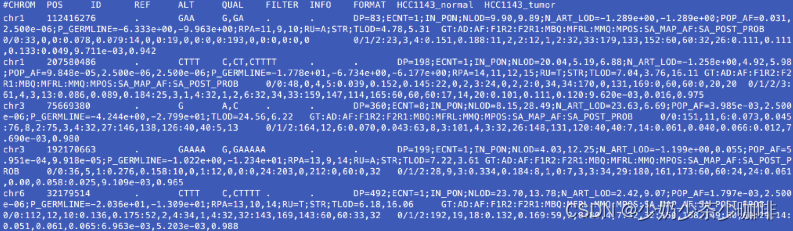
我们看到每一个变异体调用有11列信息,包括正常和肿瘤的基因型调用。注意QUAL和FILTER的空字段,以及站点(INFO)和样本水平(第10和11列)的注释。每个样本都有基因型,当一个部位是多等位基因时,我们可以看到特定等位基因的注释。样本可能有额外的注释,例如与相位有关的PGT和PID。We see eleven columns of information per variant call including genotype calls for the normal and tumor. Notice the empty fields for QUAL and FILTER, and annotations at the site (INFO) and sample level (columns 10 and 11). The samples each have genotypes and when a site is multiallelic, we see allele-specific annotations. Samples may have additional annotations, e.g. PGT and PID that relate to phasing.
1.1 What are the Mutect2 annotations?
我们可以在VCF头中查看标准的FORMAT级和INFO级Mutect2注释。We can view the standard FORMAT-level and INFO-level Mutect2 annotations in the VCF header.
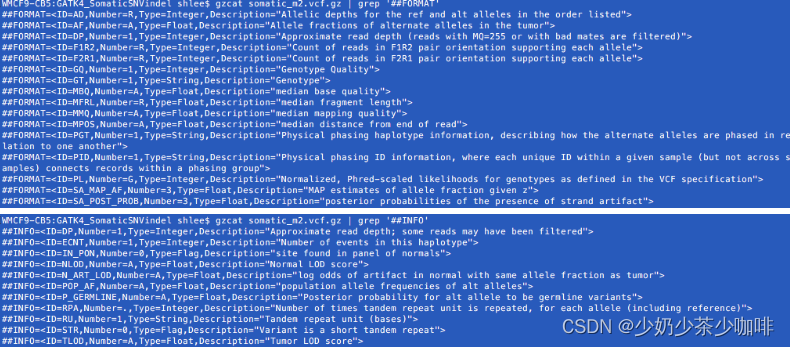
工具索引中的变体注释部分进一步描述了一些注释。关于GATK4中可用的注释的完整列表,请参见本网站。gatk/src/main/java/org/broadinstitute/hellbender/tools/walkers/annotator at master · broadinstitute/gatk · GitHub
要启用依赖于非标准注释的特定过滤,或者只是添加额外的注释,请使用-A参数。例如,-A ReferenceBases为变体调用添加了ReferenceBases注解。注意,如果过滤器所依赖的注释不存在,FilterMutectCalls将跳过特定的过滤而没有任何警告信息。To enable specific filtering that relies on nonstandard annotations, or just to add additional annotations, use the -A argument. For example, -A ReferenceBases adds the ReferenceBases annotation to variant calls. Note that if an annotation a filter relies on is absent, FilterMutectCalls will skip the particular filtering without any warning messages.
1.2 What is the impact of disabling the MateOnSameContigOrNoMappedMateReadFilter read filter?
我们利用三个种系样本做出了创建PoN的动议。这些样本是HG00190、NA19771和HG02759。
首先,在每个正常样本上以纯肿瘤模式运行Mutect2。在纯肿瘤模式下,用-肿瘤标志来分析单个病例样本,而没有附带的匹配对照-正常样本。在本教程中,我们只对HG00190样本运行此命令。
gatk Mutect2 \-R ~/Documents/ref/hg38/Homo_sapiens_assembly38.fasta \-I HG00190.bam \-tumor HG00190 \--disable-read-filter MateOnSameContigOrNoMappedMateReadFilter \-L chr17plus.interval_list \-O 3_HG00190.vcf.gz这将产生一个调用集3_HG00190.vcf.gz和一个匹配索引。Mutect2调用样本中的变体,其敏感标准与在体细胞模式下调用肿瘤中的变体相同。由于该命令省略了触发前期过滤的选项,我们希望所有可检测的变异体都被调用。调用的变体将包括低等位基因分数的变体和具有多个变体等位基因的部位,即多等位基因部位。下面是3_HG00190.vcf.gz中的两条多等位基因记录。This generates a callset 3_HG00190.vcf.gz and a matching index. Mutect2 calls variants in the sample with the same sensitive criteria it uses for calling mutations in the tumor in somatic mode. Because the command omits the use of options that trigger upfront filtering, we expect all detectable variants to be called. The calls will include low allele fraction variants and sites with multiple variant alleles, i.e. multiallelic sites. Here are two multiallelic records from 3_HG00190.vcf.gz.

我们看到对于每个位点,Mutect2都调用了参考等位基因和三个备用等位基因。GT基因型的调用是0/1/2/3。两个位点的AD等位基因深度分别为16,3,12,4和41,5,24,4。We see for each site, Mutect2 calls the ref allele and three alternate alleles. The GT genotype call is 0/1/2/3. The AD allele depths are 16,3,12,4 and 41,5,24,4, respectively for the two sites.
Comments on select parameters
这篇关于GATK4 somatic mutations analysis的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!