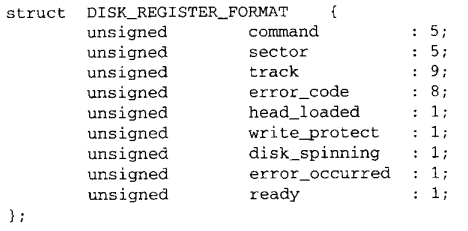
本文主要是介绍基于R和gephi做宏基因组与代谢组等多组学联合network相关性网络图,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
写在前面
拿到多组学的数据后一直在找合适的方法将二者进行关联,比如我这里是三种体液的代谢组与一种体液的宏基因组。需求是对多组学进行关联分析,直到最近看到不少文章里利用Gephi将相关性表格进行可视化的图,效果还不错,于是写个过程记录自用。这里演示的是属水平的差异菌群相对丰度(宏基因组结果)与代谢组鉴定到的差异代谢物进行关联。
主要分为两个部分:
- 先是计算相关性生成
graphml格式文件 - 使用Gephi软件进行可视化
数据准备
主要就是两个需要关联的表格
-
差异代谢物表格

-
差异菌属的相对丰度表格

这里注意:原文件如果是新建的excel,默认有3个sheet,一定记得只保留一个sheet后另存为制表符分隔的txt文件,这一点很重要,不然下面分析的时候会从读取就开始报错!

R包Hmisc计算相关性并用igraph包生成graphml
用到Hmisc包和igraph包
这里就直接放代码吧,遇到相关的报错先仔细检查数据,一般是数据格式和内容本身的问题。不行就上网找解决办法。
###微生物和代谢物的相关性网络
##计算微生物类群丰度和代谢物鉴定强度的相关系数
setwd('F:\\Analysis\\RA_Sanhe cow\\Microgenome\\Network_Gephi module_Pretreat\\RE group\\M_metabolites_genus')
library(Hmisc)
MAG <- read.delim('differ_genus.txt',row.name = 1, check.names = FALSE)
Enzyme <- read.delim('milk_differ_metabolites_inform.txt',row.name = 1, check.names = FALSE)
MAG<-t(MAG)
Enzyme<-t(Enzyme)#计算群落组成与功能的相关性,以 spearman 相关系数为例
MAG_Enzyme_corr <- rcorr(as.matrix(MAG), as.matrix(Enzyme), type = 'spearman')#相关系数 r 值和显著性 p 值矩阵
r <- MAG_Enzyme_corr$r
p <- MAG_Enzyme_corr$P#只保留微生物丰度-功能基因丰度的相关系数
#去除微生物-微生物、功能基因-功能基因之间的相关系数
r <- r[colnames(MAG),colnames(Enzyme)]
p <- p[colnames(MAG),colnames(Enzyme)]#阈值筛选
#将 spearman 相关系数低于 0.7 的关系剔除,即 r>=0.7
#该模式下,一定要注意负值的选择是否是合适的,因为很多情况下可能负相关无意义
r[abs(r) < 0.7] <- 0#选取显著性 p 值小于 0.05 的相关系数,即 p<0.05
# p <- p.adjust(p, method = 'BH') #可选 p 值校正,这里使用 BH 法校正 p 值
p[p>=0.05] <- -1
p[p<0.05 & p>=0] <- 1
p[p==-1] <- 0#根据上述筛选的 r 值和 p 值保留数据
z <- r * p#再转换为对称矩阵,igraph 只能识别这种样式的邻接矩阵类型
z1 <- MAG_Enzyme_corr$r
z1[z1 != 0] <- 0
z1[rownames(z),colnames(z)] <- z
z1[colnames(z),rownames(z)] <- t(z)#write.table(data.frame(z1, check.names = FALSE), 'MAG_Enzyme_corr.matrix.txt', col.names = NA, sep = '\t', quote = FALSE)##获得网络
library(igraph)#将邻接矩阵转化为 igraph 网络的邻接列表
#构建含权的无向网络,权重代表了微生物丰度和功能基因丰度间的 spearman 相关系数
g <- graph.adjacency(z1, weighted = TRUE, mode = 'undirected')
g#孤立节点的删除(删除度为 0 的节点)
g <- delete.vertices(g, names(degree(g)[degree(g) == 0]))#该模式下,边权重代表了相关系数
#由于权重通常为正值,因此最好取个绝对值,相关系数重新复制一列
E(g)$correlation <- E(g)$weight
E(g)$weight <- abs(E(g)$weight)#查看网络图
plot(g)#graphml 格式,可使用 gephi 软件打开并进行可视化编辑
write.graph(g, 'network.graphml', format = 'graphml')
plot包出的图

Gephi软件可视化
下载地址:The Open Graph Viz Platform,支持中文
可参考 B站:Gephi可视化进阶:教你绘制一个漂亮的网络图
进一步对上面生成的network.graphml文件用Gephi可视化
-
导入图形文件

-
导入后初始布局
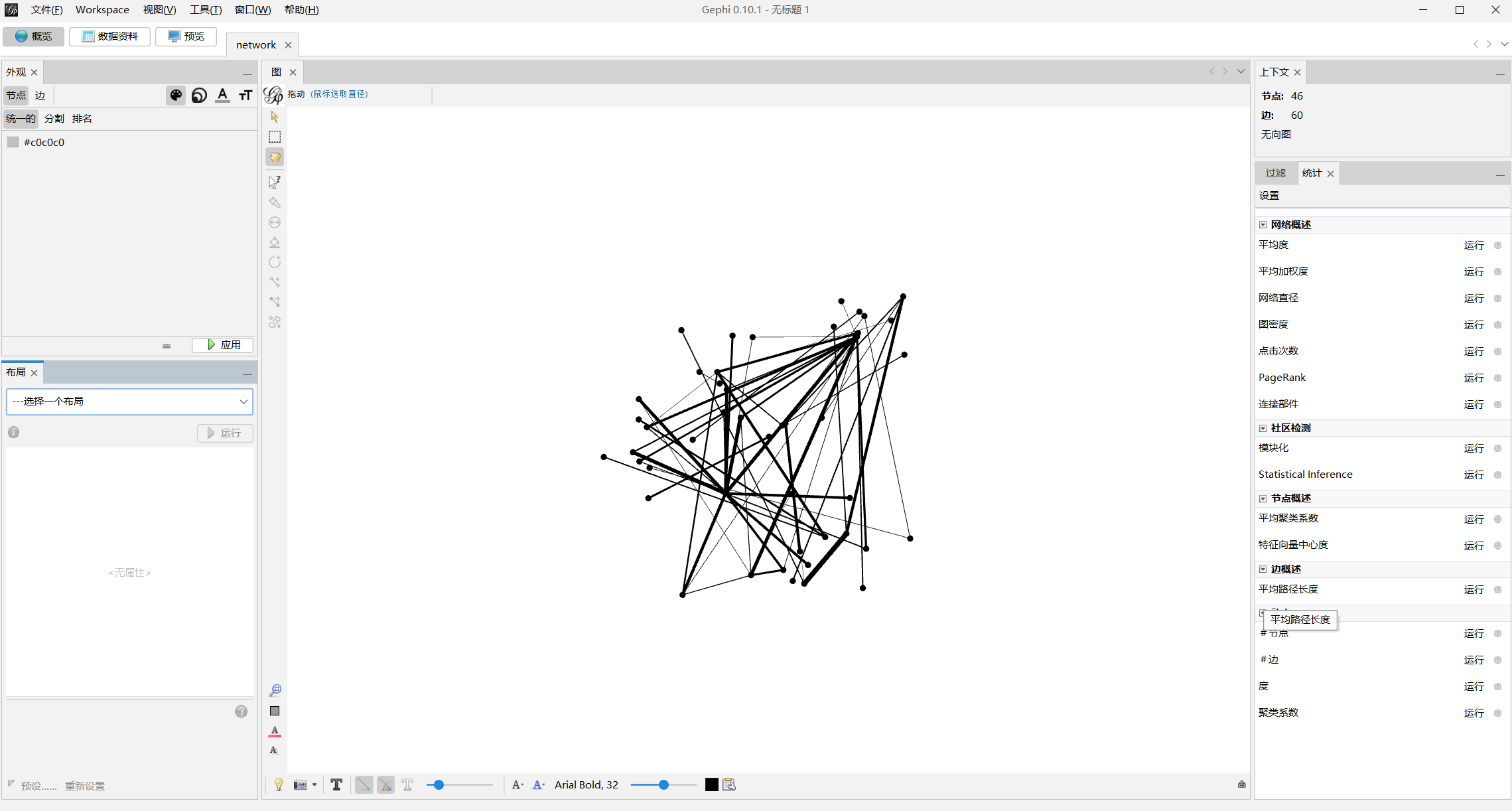
-
更改布局,等节点的位置没有移动变化即可停止

-
计算平均度与模块化

-
模块化上色

-
调整节点大小

-
添加节点标识,去界面左上角
数据资料处替换节点名字

-
预览图,这里根据需要可以自行摸索调整,红框圈出了我调整的部分

- 导出保存

效果展示
结果图细节修改:在AI中嵌入上面导出的pdf格式结果后补充图例细节即可,下方是效果展示
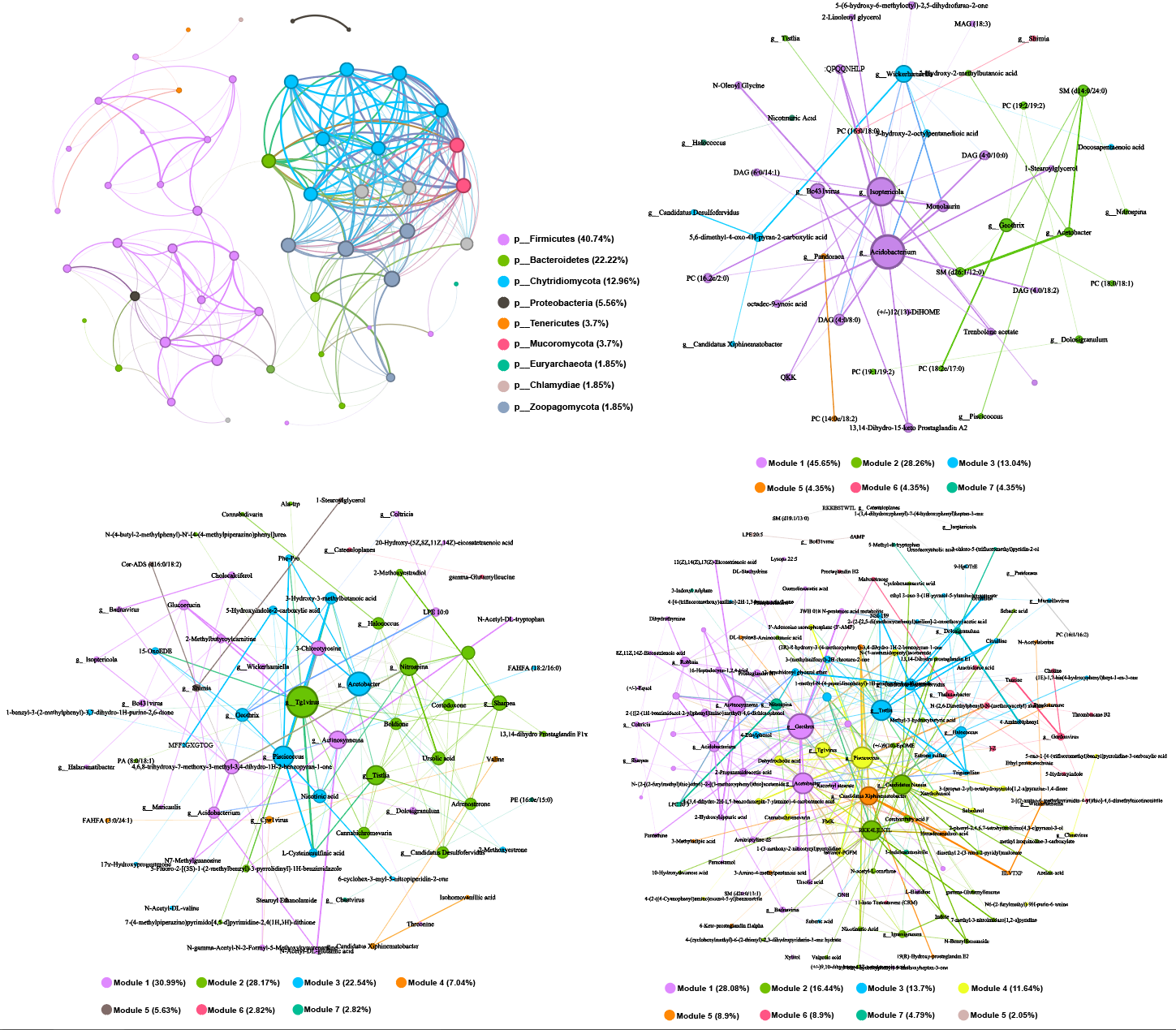
这篇关于基于R和gephi做宏基因组与代谢组等多组学联合network相关性网络图的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!