本文主要是介绍Pushing the boundaries of molecular representation for drug discovery with graph attention mechanism,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
AttentiveFP 2020
Motivations
1、The gap between what these neural networks learn and what human beings can comprehend is growing
2、Graph-based representations take only the information concerning the topological arrangement of atoms as input
3、Geometry-based representations employ the molecular geometry information, including bond lengths, bond angles, and torsional angles
4、Attentive FP automatically learns nonlocal intramolecular interactions from specified tasks
(1)characterizes the atomic local environment by propagating node information from nearby nodes to more distant ones
(2)allows for nonlocal effects at the intramolecular level by applying a graph attention mechanism
Related Works
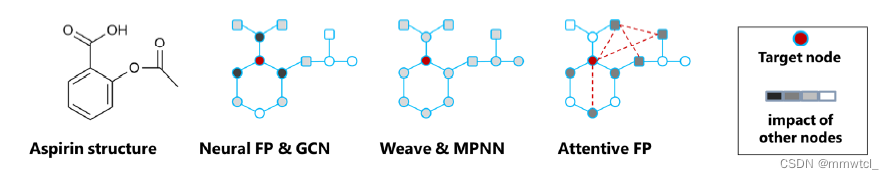
1、Neural FP and GCN
models the neighbor nodes’ chances to influence the target decrease with topological distance during the recursive propagation procedure(但有时候距离较远的结点对当前结点也会有很重要的作用)
2、Weave and MPNN
(1)construct virtual edges linking every pair of nodes in a molecule graph, meaning that any nodes, regardless of their distance to a target node, have an equal chance to exert influence, similar to the direct neighbors of the target node
(2)tended to make all the neighbors’ impacts weak because of their averaging effect
Methods
Attentive FP Network Architecture
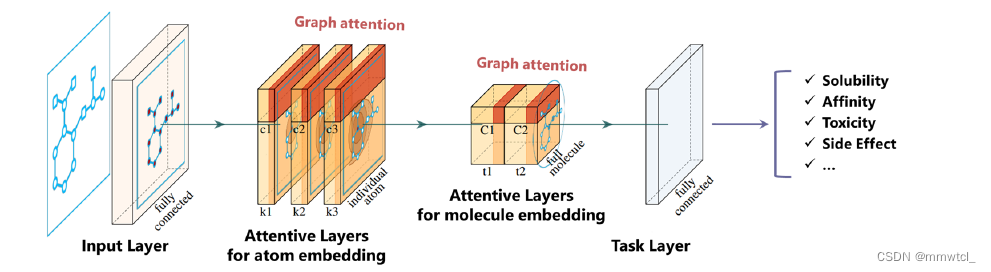
1、linear transformation and nonlinear activation were performed to unify the vector length
2、those initial state vectors are further embedded with stacked attentive layers for node embedding
3、we treat the entire molecule as a supervirtual node that connects every atom in a molecule and is embedded using the same atom embedding attention mechanism
4、The final state vector is the learned representation that encodes structural information about the molecular graph, followed by a task dependent layer for prediction
Attentive Layers on a Graph
two stacks of attentive layers to extract information from the molecular graph
1、one stack (with k layers) is for atom embedding(a single attentive layer)
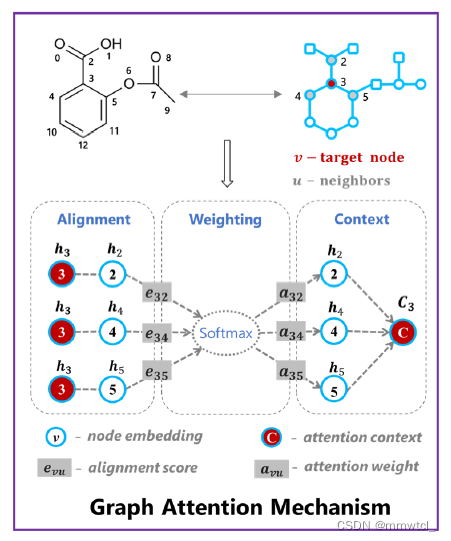
(1)When applying attention to atom 3, the state vector of atom 3 is aligned with the state vector of its neighbors 2, 4, and 5
(2)the weight that measures how much attention we want to assign to the neighbors is calculated by a softmax function
(3)a weighted sum of the neighborhood information C3 is obtained as the attention context vector of atom 3
(4)C3 (the attention context of atom 3) is fed into a GRU recurrent network unit together with the state vector h3 of atom 3(This scheme allows relevant information to be passed down without too much attrition)
2、full network architecture for the attentive layers(更新h的过程)
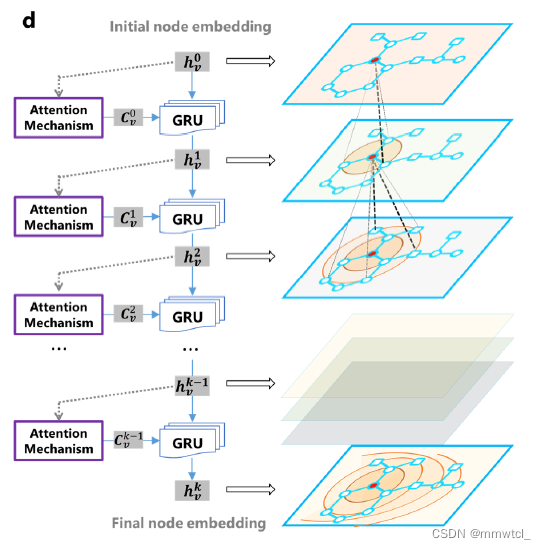
3、the other (with t layers) is for full-molecule embedding
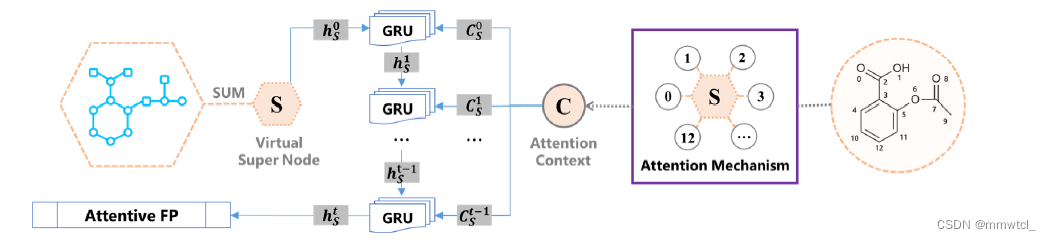
all of the atom embeddings are aggregated by assuming a super virtual node that connects all the atoms of the molecule.
Conclusions
1、The adoption of graph attention mechanisms at both the atom and molecule levels allows this new representation framework to learn both local and nonlocal properties of a given chemical structure
2、it captures subtle substructure patterns
3、inverting the Attentive FP model by extracting the hidden layers or attention weights provides access to the model’s interpretation
这篇关于Pushing the boundaries of molecular representation for drug discovery with graph attention mechanism的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!








