本文主要是介绍DeepLncPro:用于识别长非编码RNA启动子的可解释卷积神经网络模型,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
论文题目:
DeepLncPro: an interpretable convolutional neural network model for identifying long non-coding RNA promoters
期刊:Briefings in Bioinformatics
论文链接:https://doi.org/10.1093/bib/bbac447
代码链接: https://github.com/zhangtian-yang/DeepLncPro
背景
长链非编码RNA (Long non-coding RNA, lncRNA)是一种长度大于200个核苷酸的非编码RNA。启动子是位于转录起始位点(transcription start site, TSS)上游的调控元件,通过与转录因子的结合,启动并调控RNA的转录。
本文提出了一种基于卷积神经网络(CNN)的方法,称为DeepLncPro,在人和小鼠中识别lncRNA启动子。在DeepLncPro中,利用单核苷酸化学性质和二核苷酸物理化学性质对序列进行编码。为了获得鲁棒模型,对CNN进行超参数优化,获得最优超参数。
数据
来源:在真核启动子数据库(Eukaryotic promoter Database, EPD)中获得人和小鼠lncRNA启动子序列
特征表示
one-hot
将' A '编码为(1,0,0,0),' T '编码为(0,1,0,0),' G '编码为(0,0,1,0),' C '编码为(0,0,0,1)。因此,长度为L的DNA序列可以转化为4 × L矩阵A1.
NCP
这四种脱氧核糖核苷酸携带不同的碱基,在环结构、氢键强度和化学功能上存在差异。在环的数量方面,“A”和“G”包含两个环,而“C”和“T”包含一个环。在氢键强度方面,“a”和“T”之间形成弱氢键,而“C”和“G”之间形成强氢键。就化学成分而言,‘A’和‘C’属于氨基而‘G’和‘T’属于酮族。因此,' A '被编码为(1,1,1),' T '被编码为(0,1,0),' G '被编码为(1,0,0),' C '被编码为(0,0,1)。
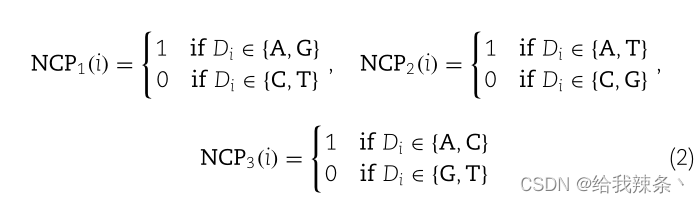
I是脱氧核苷酸在序列中的位置;NCP1、NCP2、NCP3分别代表三种化学性质。利用NCP可以将长度为L的DNA序列转化为3 × L的矩阵A2。
DPCP
连续的脱氧核苷酸组合具有不同的物理化学性质,这是基因组功能元件鉴定[30]的重要特征,已被用于启动子预测。本研究使用twist、tilt、roll、shift、slide、rise 6个DPCP编码DNA序列。
它们的值是从之前的工作中获得的。由于其值在不同的范围内变化,采用最小-最大归一化方法将其缩放到[0,1]的范围内。基于这6个DPCP,长度为L的DNA序列可以转换为6 × (L−1)矩阵。为了确保矩阵的列数与矩阵A1和A2的列数相同,使用滑动二聚体窗口算法计算每个核苷酸的DPCP。
其中DPCPn(i)表示第i个核苷酸的第i个物理化学性质,Xn表示第n个(n = 1,2,…,6)二核苷酸理化性质,分别以前二聚体Di−1Di和后二聚体DiDi + 1为输入。两个末端核苷酸D1和DL分别只依赖于两端二核苷酸的数据。据此,得到了一个6 × L的矩阵A3,该矩阵描述了该序列的理化性质。
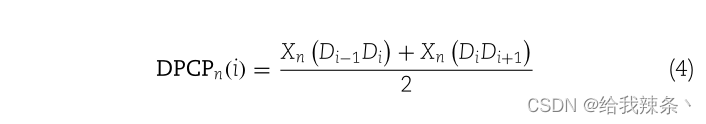
模型
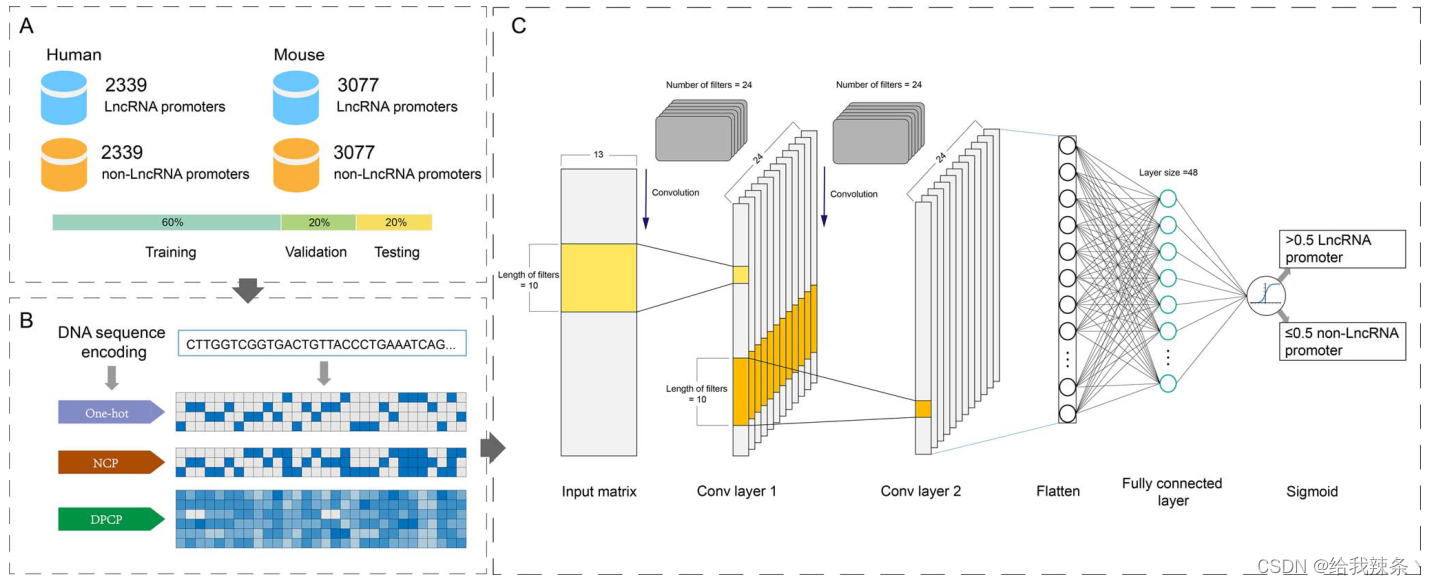
DeepLncPro概述:
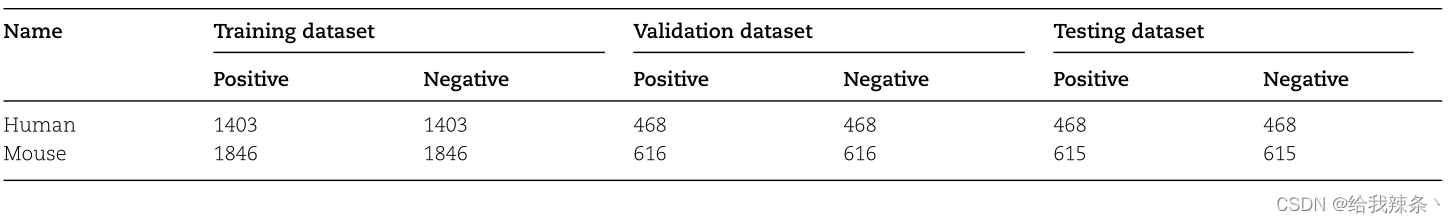
(A)数据集。该数据集包含2339个人类阳性和2339个阴性样本,3077个小鼠阳性和3077个阴性样本。每个样品在61 bp至301 bp的不同长度处截取,步长为40 bp。
(B)特征编码。使用三种特征编码方法对这些样本进行编码。编码后的特征被合并到一个13 × L矩阵中。
(C) DeepLncPro的框架。DeepLncPro基于卷积神经网络构建。每个样本都有一个预测分数,范围从0到1。如果评分为> 0.5,则预测该序列为lncRNA启动子;否则是非lncrna启动子。
超参数优化与模型选择
为了获得性能和泛化能力更好的模型,我们进行了超参数优化。为了使训练过程更加稳定,采用Adam算法基于批量梯度下降自动确定学习率。采用随机搜索方法确定学习速率、神经元数量、卷积层大小和滤波器数量等超参数。
在超参数优化过程中,我们首先通过在合理范围内选择一组超参数来训练一个基本模型。然后,通过保持其他超参数不变,在给定范围内搜索某个超参数。根据从验证数据集中获得的性能,选择最优超参数。重复这个过程,直到所有超参数都被优化。一旦确定了所有超参数,它们就被用于在训练和验证数据集上再次训练DeepLncPro。需要指出的是,只保留了验证集中精度最高的超参数组合。
绩效评估
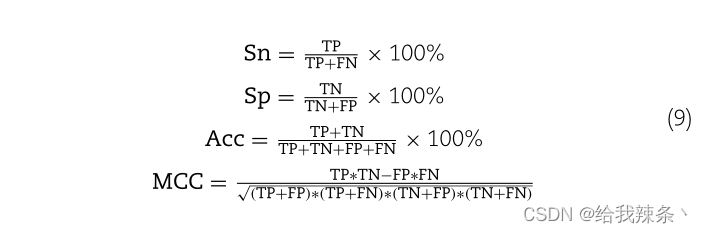
结果与讨论
序列长度和编码方案对模型的影响
为了确定预测lncRNA启动子的最佳序列长度和编码方案,研究了序列长度和编码方案对模型性能的影响。为此,我们基于不同类型的序列长度和编码方案的组合建立了不同的模型。为了得到具有良好泛化能力的模型,将人和小鼠的训练数据结合起来进行训练。对每个模型的超参数,按照超参数优化与模型选择章节介绍的步骤进行优化。
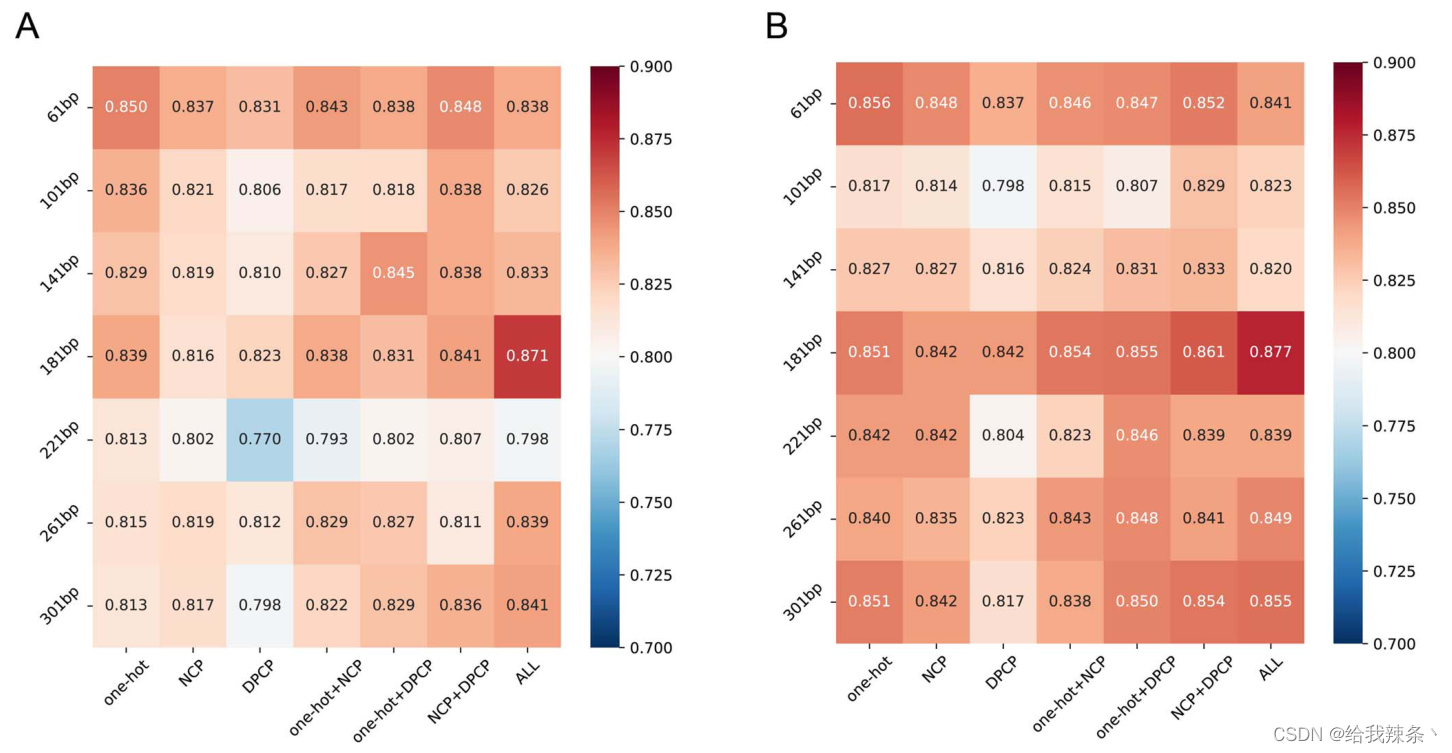
基于不同序列长度和编码方案的模型性能。纵坐标表示从61到301 bp的序列长度。横坐标表示不同的编码方案,包括one-hot、NCP、DPCP及其组合。(A)不同模型识别人类lncRNA启动子的预测准确性;(B)不同模型在小鼠中鉴定lncRNA启动子的预测准确性
与经典机器学习方法的比较
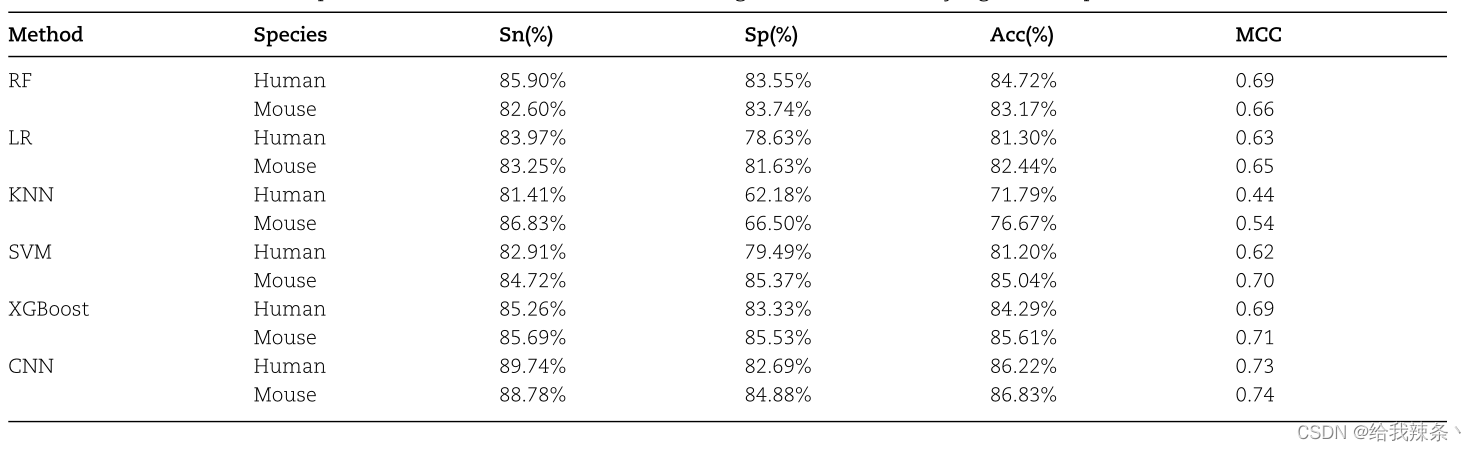
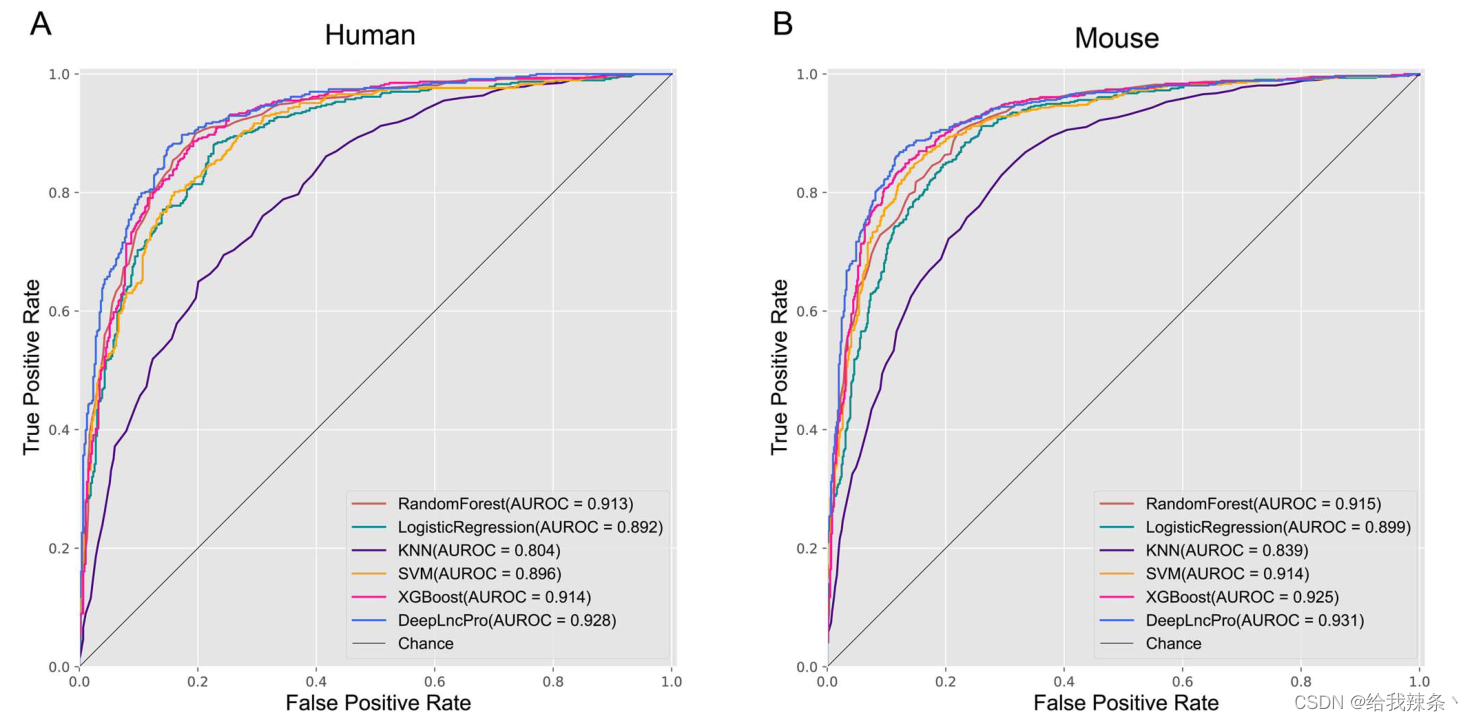
考虑到机器学习方法在DNA序列元素识别中被广泛应用,我们将DeepLncPro与随机森林(RF)、逻辑回归(LR)、k-最近邻(KNN)、支持向量机(SVM)和极端梯度增强(XGBoost)这五种经典ML方法进行了比较。将DeepLncPro的三个输入矩阵化为一个13 x L维向量,分别作为RF、LR、KNN、SVM和XGBoost的输入。DeepLncPro和ML模型用于识别测试数据集中人类和小鼠lncRNA启动子的评估指标如表2所示。DeepLncPro在识别人和小鼠lncRNA启动子方面的准确率最高,分别为86.21%和86.82%。我们还在图3中绘制了DeepLncLoc和机器学习方法的ROC曲线。结果发现,DeepLncPro在预测人类和小鼠lncRNA启动子方面的auc分别为0.928和0.931,优于其他机器学习模型。
与现有预测器的比较
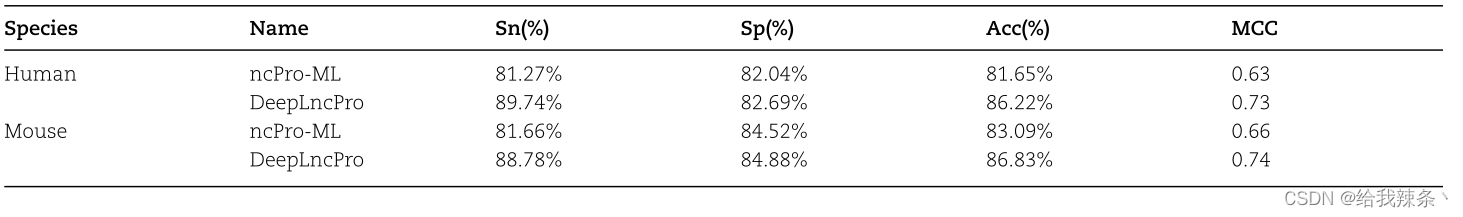
为了进一步说明其优越性,我们将DeepLncPro与现有的预测器ncPro-ML进行了比较。为了进行公平的比较,这两个预测器都在同一个测试集中进行了验证。如表3所示,DeepLncPro识别人、小鼠lncRNA启动子的准确率分别比ncPro-ML高4.57%和3.74%。相应的敏感性、特异性和Matthew’s相关系数在人组分别提高了8.47%、0.65%和0.10,在小鼠组分别提高了7.12%、0.36%和0.08。这些结果表明,DeepLncPro在鉴定人和小鼠lncRNA启动子方面更有优势。
模型解释与可视化
为了解释所提出的模型的性能,我们提取并可视化了DeepLncPro所有层的输入和输出,即原始输入、第一卷积层的输出、第二层卷积层的输出和全连接层的输出。为了便于理解这些特征,我们使用UMAP表示阳性和阴性样本的分布。结果发现,在原始输入特征形成的特征空间中,正样本和负样本无法分离(图4A)。然而,基于第一和第二卷积层的输出特征,正样本和负样本之间的边缘在特征空间中分离得更加清晰(图4B和C)
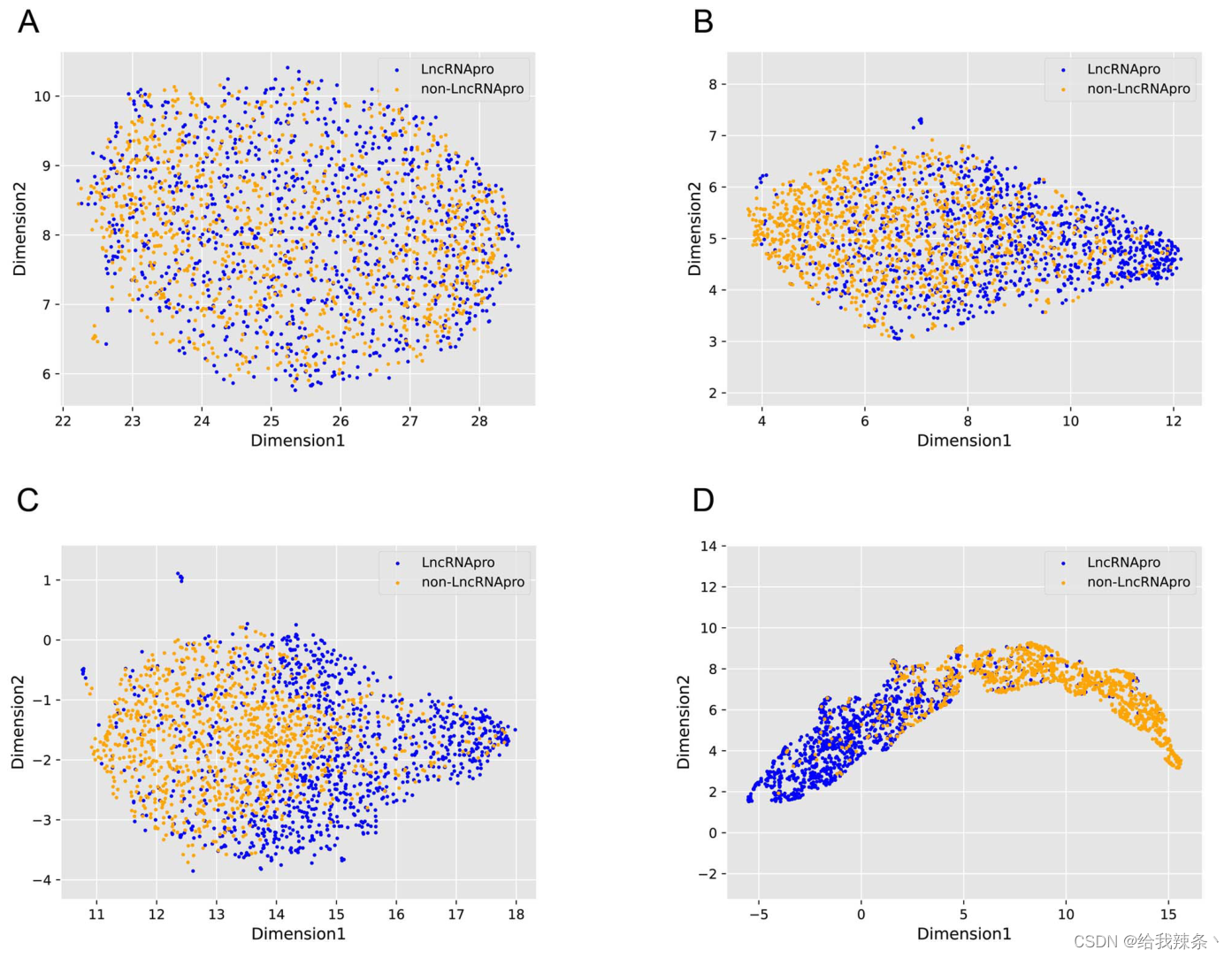
正、负样本在二维特征空间中的分布。蓝色和橙色的圆点分别代表阳性和阴性样本。(A)原始输入特征的特征空间。(B)基于第一卷积层输出的特征空间。(C)基于第二卷积层输出的特征空间。(D)基于全连接层输出的特征空间
这篇关于DeepLncPro:用于识别长非编码RNA启动子的可解释卷积神经网络模型的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!





