本文主要是介绍「杂谈」Nanopore组装的拟南芥基因组效果如何?,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
使用的数据来自于一篇发在NC的拟南芥的基因组文章,文章用了minimap/miniasm 进行组装,然后用racon和Pilon进行polish, 最后拼接处62 contigs 且N50 = 12.3 Mb。
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR217/003/ERR2173373/ERR2173373.fastq.gz
seqkit seqkit fq2fa ERR2173373.fastq.gz | gzip -c > ERR2173373.fasta
我用的是Canu进行组装,参数如下
canu \-p ath -d Athaliana\useGrid=true \gridOptions="-S /bin/sh -q wangjw" \gridEngineArrayMaxJobs=20 \gridEngineThreadsOption="-pe openmpi THREADS" gridEngineMemoryOption="-l mem_free=MEMORY" \minReadLength=2000 maxThreads=15 maxMemory=60G \genomeSize=100m \rawErrorRate=0.300 \correctedErrorRate=0.100 \-nanopore-raw ERR2173373.fasta.gz
Canu默认Pacbi的rawErroRate是0.300, Nanopore是0.500。但是根据我在自己建立的基因组学群里的讨论,目前nanopore的单条read的错误率大概是12%,所以两条read在overlap的时候,最差估计会有24%以上的序列差异,于是我尝试设置了0.300. 但是由于Nanopore的错误率不是完全随机(经群里的小伙伴告知),所以纠错后正确率低于Pacbio, 所以我设置了0.100. 其他参数没有修改, 最终我拼出了360条contig,N50=4.45M。
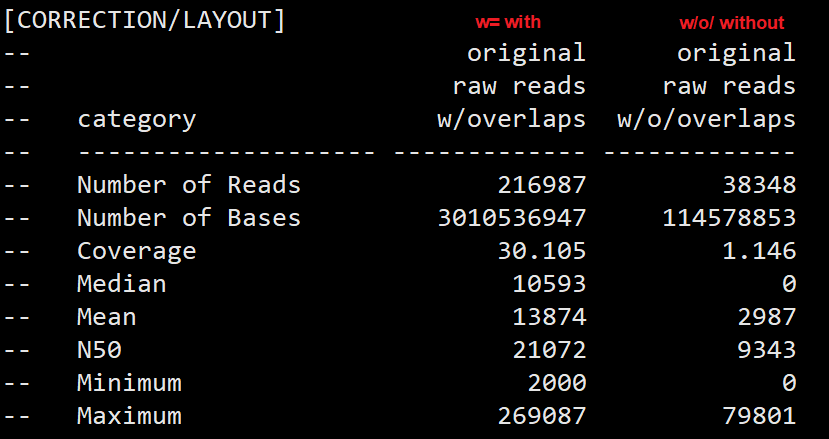
我检查了下最后输出的report文件. 第一部分表明,大部分的reads都是能够overlap。
Part II 关于多少数据用于纠错,以及预期留下多少数据。默认Canu只选择最长的40X进行纠错,可以用corOutCoverage=100调整成100X. 注: rescued 表示的是剩下的没有用于纠错的read,他们可能是质粒、线粒体等。Canu保留的目的是为了避免在组装时缺失序列信息。
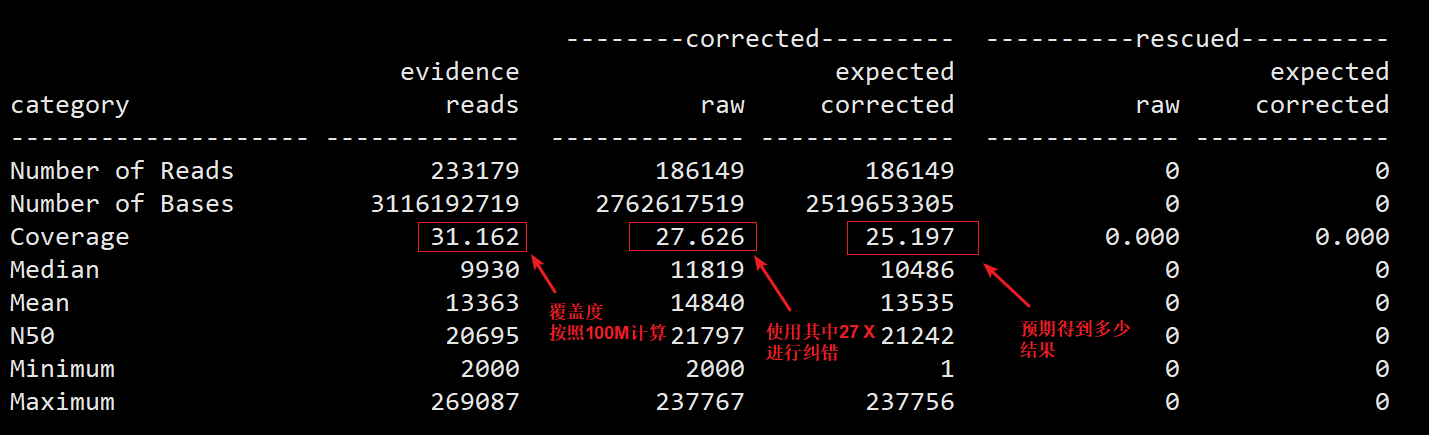
Part III: 省下的就是由于太短,不能用于纠错的部分。

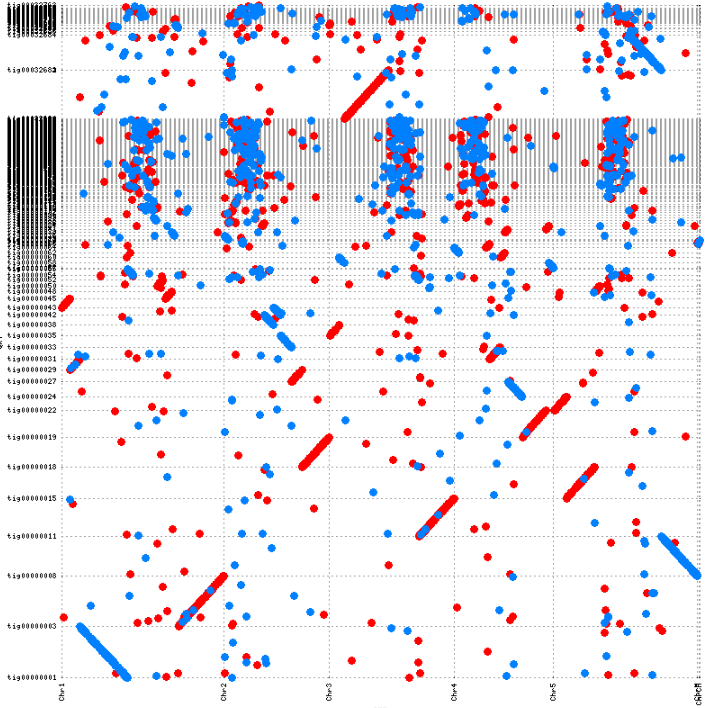
最终结果,我还用MUMMER分析了以下共线性,代码如下,
nucmer -t 20 --prefix ont2ath Athaliana.fa ath.contigs.fasta
mummerplot -p ont2ath ont2ath.delta --png --filter
基本上每条contig都主要和一条染色体存在很好的共线性,不存在contig的mis-assembly(错误组装)现象。
下一步的计划
- 只Correction 不Trim 直接组装,比较组装效果
- 提高纠错前的错误率,保持纠错后的0.1错误,比较组装效果
- 保持纠错前的错误率,提高纠错后的错误率,比较组装效果
这篇关于「杂谈」Nanopore组装的拟南芥基因组效果如何?的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!