vasp专题

linux ubuntu12.04 下的 vasp 5.2 的安装方法
#ifort:parallel_studio_xe_2013_update2,可在官网:software.intel.com/en-us/articles/non-commercial-software-download/下载,或者在百度网盘下载:http://pan.baidu.com/s/1o6sPd8m 。 #Intel 的 MKL #VASP 5.2.2 ###############
【VASP学习】在Ubuntu系统安装vasp.5.4.4的全过程(包括VASP官方学习资料、安装过程中相关编辑器的配置、VASP的编译及VASP的测试)
在Ubuntu系统安装vasp.5.4.4的全过程 VASP的简介与相关学习资料安装前的准备工作及说明安装过程intel编译器的安装VASP的编译VASP的测试 参考来源 VASP的简介与相关学习资料 VASP(Vienna Ab initio Simulation Package)是基于第一性原理对原子尺度的材料进行模拟计算的软件。比如可以进行原子尺度材料的电子结构计算、分子

VASP笔记之:计算德拜温度,杨氏模量,弹性矩阵
VASP笔记之:计算德拜温度,杨氏模量,弹性矩阵 最近需要计算杨氏模量,但是上面三个量都是一起算出来的,so,一起记录一下笔记。 使用版本为VASP5.4.4,为了方便计算使用的是vaspkit1.2.1软件辅助自动生成的脚本进行的计算,微信公众号:学术之友,原文链接为:这里 下面我们以金刚石结构为例讲解如何采用应力-应变函数关系计算弹性常数,详见VASPKIT/examples/elast

【VASP解读】Nat. Commun.:CoFe@FeOx助力Li/Na-S电池
均匀分布在载体上的复杂金属纳米颗粒具有独特的物理化学性质,具有广泛的应用前景。常用的湿化学方法在同时实现纳米颗粒结构设计和均匀分散方面存在局限性,但固相合成可在载体上制备复杂的金属纳米颗粒。基于此,中国科学院大连化学物理研究所刘健研究员、中国科学技术大学余彦教授等人报道了利用固相合成策略,精确合成分布均匀的CoFe@FeOx核-壳纳米颗粒。CoFe@FeOx NPs调控多硫化物的双功能
VASP结合vaspkit+ShengBTE计算热电优值(一)
电导率σ,塞贝克系数S的计算: 使用vaspkit计算处对应的物理量,具体流程为: 准备好计算的材料对应的POSCAR。如果是二维材料可以使用vaspkit 的921或923功能对二维材料POSCAR进行标准化。进行结构优化。使用 vaspkit-681命令生成高密度的KPOINTS,然后进行静态计算 (注意只有使用这项功能生成KPOINTS计算的结果才能继续使用vaspkit命令计算下一步,使用

linux超线程会拖慢vasp计算速度,VASP跑MD的问题
17 个回复 willykohn 耳朵 2008-04-16 1. 1000步太少了。 2. 初始态离平衡态太远了 【 在 abalone (纳米光学?) 的大作中提到: 】 : 想在300K下跑一个NVE : 于是先把系统在300K下跑了1000步NVT : 然后用了两种方法跑NVE : ................... abalone 纳米光学? 2008-04-16 初始态是0K
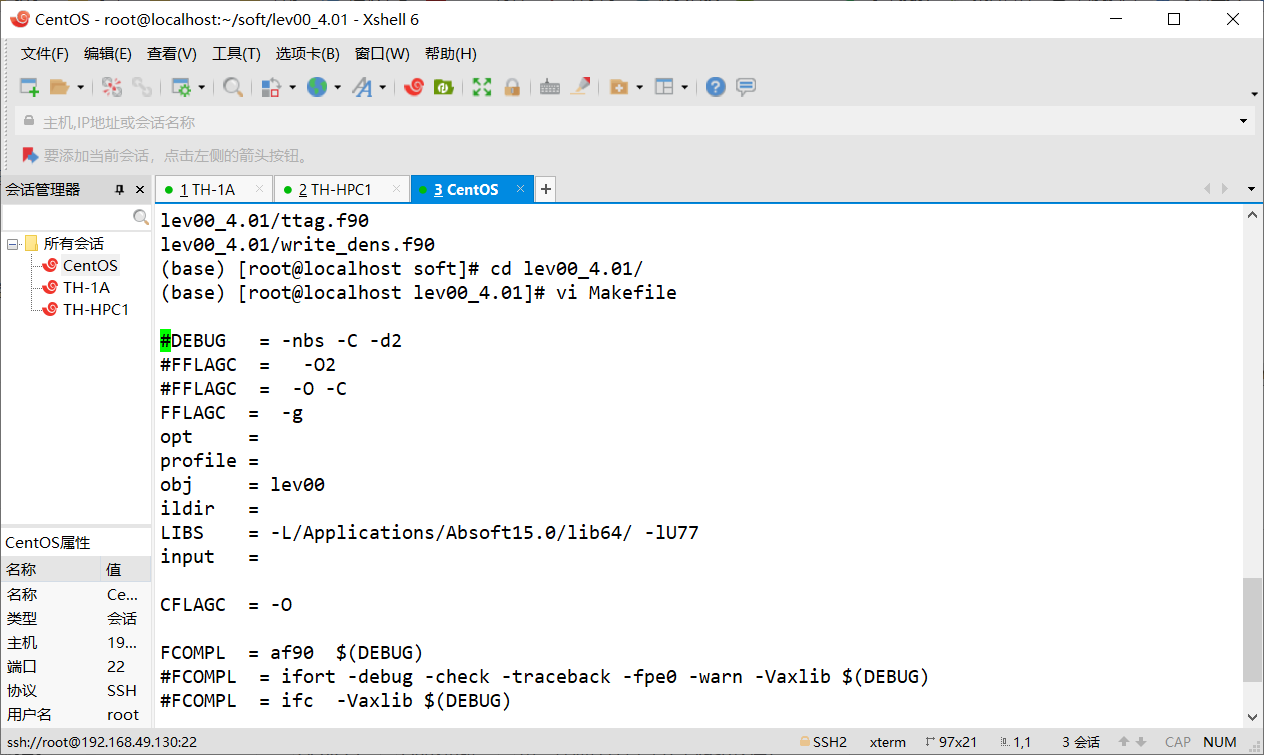
vasp-Lev00编译
LEV00 - a useful tool for various DFT codes (VASP, SIESTA, CASTEP, QUICKSTEP) Lev00 虽然有很多不同的功能,但是我们一般只用它处理电荷密度,来生成更容易理解的科研绘图。 例如: 如何安装LEV00? 下载LEV00,点击lev00即可下载。下载链接为:https://nms.kcl.ac.uk/l

VASP计算中的问题2----关于SOC计算报错
问题 VASP提供SOC(自旋轨道耦合)的计算,针对与重原子的相对论效应,不过SOC计算需要非共线版本的VASP,若编译的VASP为共线版本,计算SOC时会报错. 解决方案 若出现此报错,首先要意识到是编译文件的问题,需要删除当前VASP,修改makefile文件重新编译VASP. 仅仅是vasp.5.3文件夹下的问题,所以比较好处理, cp /opt/vasp/vasp.5.3/make
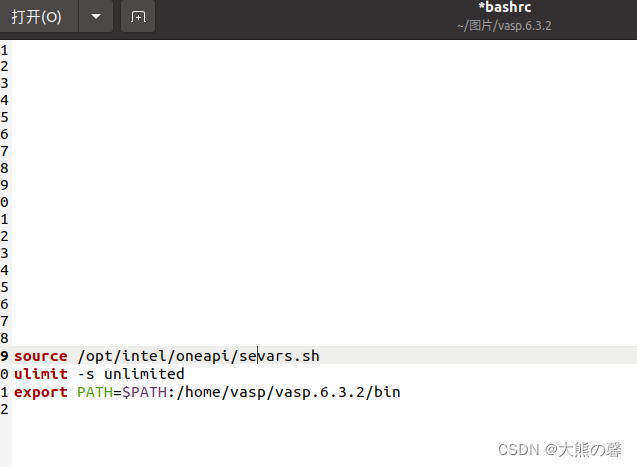
vasp安装,vasp一键安装,vasp6.4.1安装,vasp6.3.2安装,vasp5.4.4安装
1、准备由本人开发的一键安装材料,支持ubuntu18,20,22,centos7等系统,具体如下: 2,打开一个终端,如ubuntu20.04系统,执行sudo su,获取root权限,执行./intel.sh,一键式自动部署vasp所需安装环境。 3,上述终端,输入exit退出root环境,执行./vasp.sh,一键式自动编译vasp,支持intel和a

计算raman光谱,vasp+phonopy+phono3py+Phonopy-Spectroscopy
计算raman光谱,vasp+phonopy+phono3py+Phonopy-Spectroscopy 一、计算raman光谱软件介绍二、phonopy、phono3py、Phononpy-Spectroscopy安装三、raman谱计算1、高精度优化2、phonopy扩胞3、计算二阶力常数4、获得mesh文件5、确定具有raman活性的声子模式6、计算三阶力常数7、计算线宽8、电导常数计

vasp+phonopy-QHA计算材料热膨胀系数与格林奈森常数等脚本
脚本分享 通过使用QHA方法可以计算得到材料的热膨胀系数,热容和格林奈森参数等数据,这里使用的软件为vasp和phonopy。通过脚本一次计算得到相关数据。 相关参考为: https://mp.weixin.qq.com/s/dC7btVnmTxwwhqCJpL8YvQ http://phonopy.github.io/phonopy/examples.html
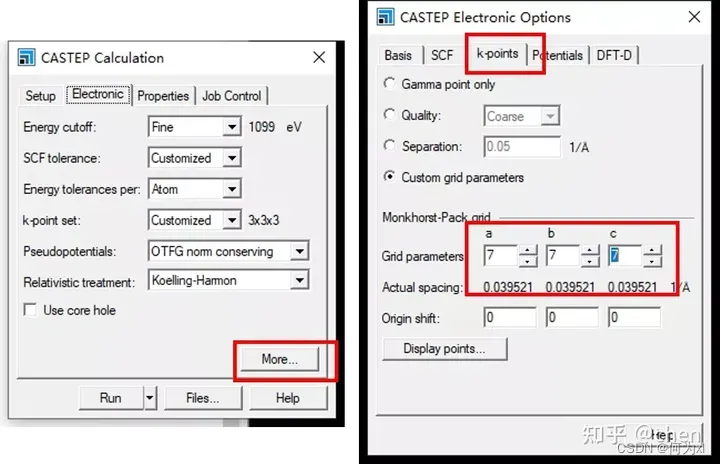
【VASP】KPOINTS文件介绍
【VASP】KPOINTS文件介绍 一、KPOINTS 的两种结构第一种结构:(非对称)第二种结构:(高对称) 二、关于KPOINTS设置的一些经验三、KPOINTS的选取 前言 一、4个常用的输入文件INCAR、POSCAR、POTCAR、KPOINTS INCAR: 计算任务类型是什么?怎么计算? KPOINTS: 包含了倒易空间点网格的坐标和权重。 POSCAR: 包含元

【VASP】POSCAR文件
【VASP】POSCAR文件 前言 一、4个常用的输入文件INCAR、POSCAR、POTCAR、KPOINTS INCAR: 计算任务类型是什么?怎么计算? KPOINTS: 包含了倒易空间点网格的坐标和权重。 POSCAR: 包含元胞的原子坐标信息以及初始速度等信息。 POTCAR: 超软赝势或PAW势函数(有一个赝势库)。 POSCAR文件:位置文件。描述所计算体系

VASP结构优化(1)——分子的优化
本文转载于微信公众号,VASP学习交流,将会持续更新 分子和团簇的优化 构建大的supercell,在考量计算量的情况下,保证近邻image中最近原子距离>6A (可以认为破坏对称性) 只优化原子坐标,不优化晶格常数:VASP中ISIF=2, K点也是单Gamma点(见KPOINTS详解) 分子H2O的建立POSCAR 1. 从菜单栏选择File —— New,或者直接点击如图中的

VASP新手入门,对于VASP以及Linux系统初学者的福音~(附VASP简单结构优化的详细过程)
其实好多朋友们对于突然被丢过来一个课题,去学习VASP是完全没有概念的,例如什么是VASP?VASP是一个什么样的软件?(好多的同学们在找我帮忙编译安装过VASP之后最有趣的一句话是“您好!请问VASP这个软件在哪里,我为什么找不到!”)如何使用VASP?用VASP到底去计算什么?等等等,那么我就来简单的,特别小白通俗易懂的介绍一下这个貌似很是高深其实很好上手的维也纳从头算(VASP).