本文主要是介绍VASP结合vaspkit+ShengBTE计算热电优值(一),希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
- 电导率σ,塞贝克系数S的计算:
使用vaspkit计算处对应的物理量,具体流程为:
- 准备好计算的材料对应的POSCAR。如果是二维材料可以使用vaspkit 的921或923功能对二维材料POSCAR进行标准化。
- 进行结构优化。
- 使用 vaspkit-681命令生成高密度的KPOINTS,然后进行静态计算 (注意只有使用这项功能生成KPOINTS计算的结果才能继续使用vaspkit命令计算下一步,使用M-S方法自动生成K点的计算结果无法进行下一步)。
- 准备对应的http://INPUT.in文件用于输运性质计算

关于INPUT文件中的参数,一般只需要调整Temperature 以及Relaxation time。其中Relaxation time 一般可以通过查找文献得到当前研究的结构的载流子Relaxation time/scattering time 。当我们查找不到时就需要自己计算出Relaxation time。
- 晶格热导率的计算
计算晶格热导率我们需要用到的软件包括Phonopy,Thirdorder,ShengBTE。
其中Phonopy用于计算声子谱及二阶力常数,Thirdorder用于计算三阶力常数,ShengBTE用于结合前面两者的结果计算晶格热导率。
1、Phonopy计算声子谱及二阶力常数
计算声子谱及二阶力常数的具体流程如下:
(1)对初始结构进行高精度的结构优化
这一步中INCAR的主要参数是EDIFFG,一般情况下应达到EDIFFG=-1E-8的标准。考虑优化速度,可以通过优化多次,每次优化时逐步减小EDIFFG直到EDIFFG=-1E-8的方法进行优化。高精度优化中IBRION建议设置为1,且当EDIFFG较小时建议设置ISIF=2。
(2)使用Phonopy进行扩胞
一般情况下,扩胞后的超胞中的原子数达到100就可以了。扩胞后会生成SPOSCAR 与 POSCAR-* 等文件。前者可用于DFPT(密度泛函微扰)方法,后者应用于有限位移法。两种方法计算的结果没有区别。

(3)DFPT法
DFPT法需要使用SPOSCAR进行计算(单个任务)。可参考王宁博士在B站的视频[7]。笔者在这里贴出自己的代码仅供参考。
计算前需要将原高精度优化的POSCAR 重命名为POSCAR-unitcell SPOSCAR命名为POSCAR。

vasp计算完成后编写band.conf 并运行以下命令:


这样就能得到二阶力常数文件FORCE_CONSTANTS,以及声子谱的图band.pdf数据a.dat。对于声子谱,我们要保证没有虚频,这样才能保证晶格的稳定性。
2、ThiRDoRDer计算三阶力常数
首先需要对高精度优化后的结构扩胞:
![]()
与phonopy不同的是参数-d表示考虑哪些近邻原子的受力来计算力常数矩阵。d为正数时表示截断半径(单位nm) 为负数时表示所考虑近邻原子的个数。
一般情况d取<=-10 或 >=0.6。 这会影响到扩胞生成的POSCAR-*的个数与计算精准度。
这里使用有限位移法进行计算。
首先准备INCAR,POTCAR,KPOINTS

然后用脚本生成文件夹:

通过脚本提交作业:
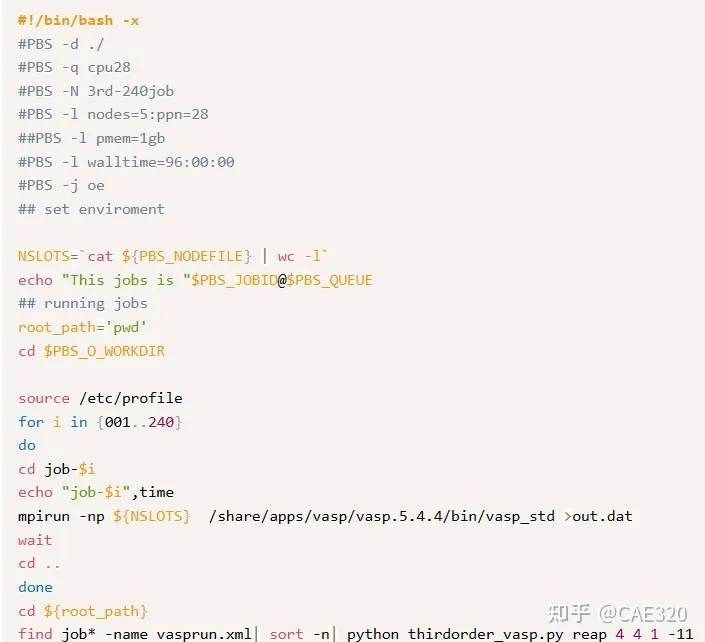
最后就能得到三阶力常数矩阵文件FORCE_CONSTANTS_3RD。这一步一般需要非常长的时间去进行计算,因此扩胞的大小可以稍微比二阶力常数矩阵的计算时的大小小一点。
这样我们就得到二阶力常数矩阵与三阶力常数矩阵
最后,有相关需求欢迎通过公众号“320科技工作室”联系我们
这篇关于VASP结合vaspkit+ShengBTE计算热电优值(一)的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!






