biostar专题
biostar handbook(七)| BLAST
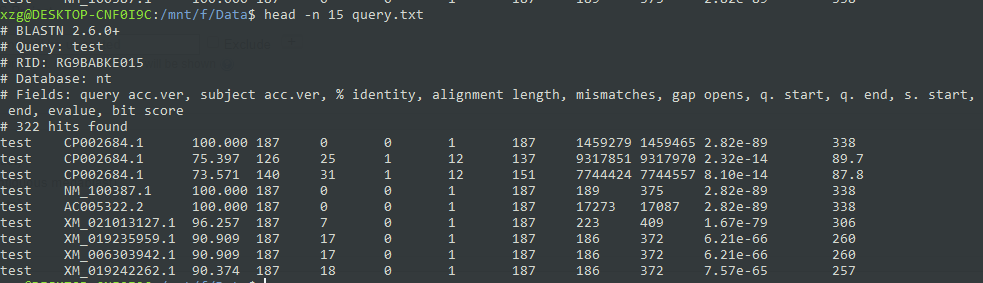
Basic local alignment search tool (BLAST) 包括:blastn, blastp, blastx, tblastn, tblastx等. 使用conda安装即可。 conda install -c bioconda blast# blast安装perl模块的方法conda isntall perl-digest-md5 BLAST的主要理念 Se
biostar handbook(六)|正则表达式和K-mers在模式匹配中的使用
模式匹配中的正则表达式和k-mer 模式匹配指的是在看似杂乱无章的系统中找到符合要求的部分。比如说你想从基因组中寻找motif,转录因子结合位点,CDS, 或者检测测序结果里是否有接头等,这些行为都可以解读为根据已有的模式去寻找目标序列中符合要求的片段。后面介绍的序列模式匹配的两种方法,正则表达式和k-mers。 正则表达式的基础概念 正则表达式(regular expression)的概念

biostar handbook: 第五周笔记汇总+第六周任务布置
简单总结下第五周的笔记: biostarhandbook(五)|序列从何而来和质量控制YXF-测序仪和质量控制Biostar学习笔记(5)测序平台、测序原理及质量控制Biostar第六课 测序仪和QC 顺便发布第六周的任务: 第六周的任务是第十章和第十一章。其中第十一章介绍的是序列模式,是对正则表达式的温故知新。第十一章则是开始序列分析的重要一步:序列联配。任务如下: 什么是正则表达式什么
biostar handbook(五)|序列从何而来和质量控制
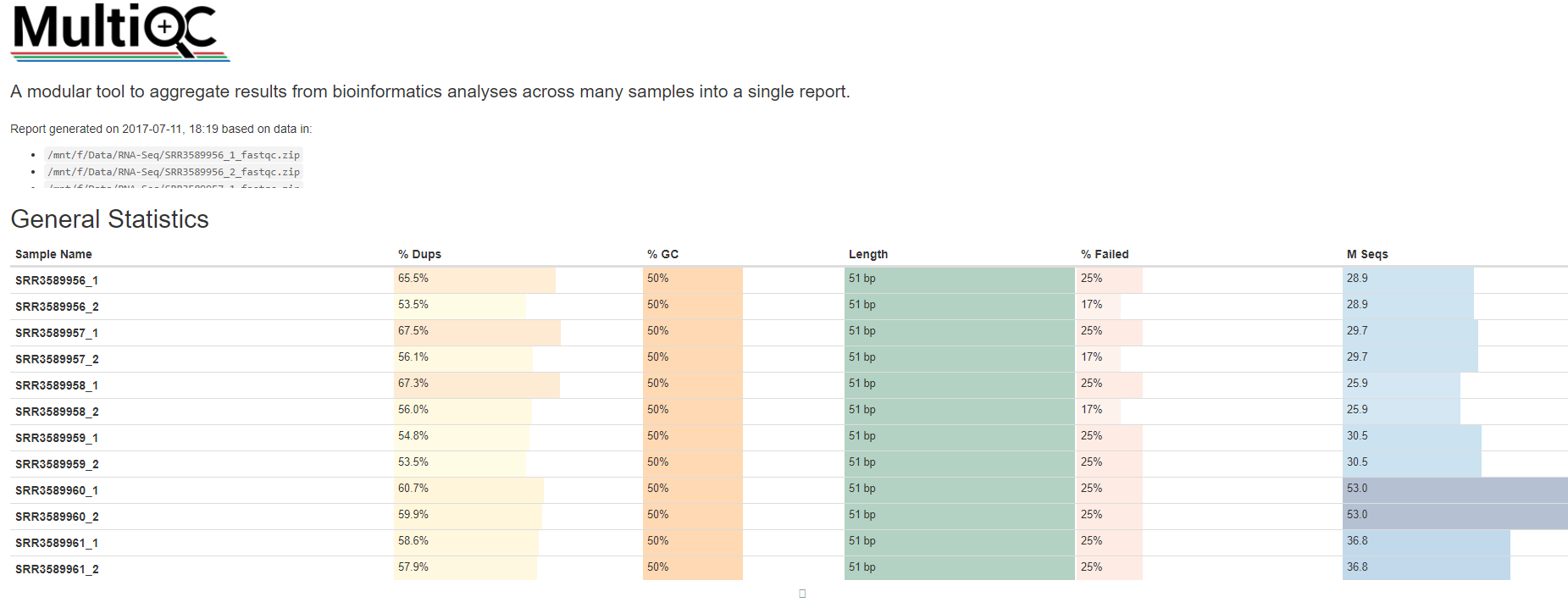
测序仪 2017年一篇发表在Nature的综述"DNA sequencing at 40: past, present and future"介绍了DNA测序这40年的发展历程。1976年,Sanger和Coulson同时发表了2种方法用于对上百个DNA碱基进行解码,这就是第一代测序技术。到了2005年,罗氏的454平台揭开了高通量测序的序幕,后面则是SOLiD,454和Illumina三方对抗

biostar handbook: 第四周笔记汇总+第五周任务布置
不知不觉已经过去了四周,这个系列的开篇语写于2017年10月14日,距离今天差不多是一个月的时间了。这个月的时间学的内容并不算多,大致也就是如下几个内容: *nux基础: 这个尤为重要,学会了*nix(Linux或unix)之后,如果能够在日常科研生活中进行使用,那么你的效率将会大大提高。数据格式: 数据的保存具有一定的格式,处理数据的前提在于知己知彼,这样才能选择合适的工具。这个部分内容包括知
biostar handbook(四)|生物数据及其下载和基本操作
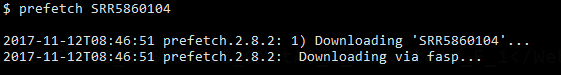
2017/11/9 第一版: 生物数据库,基本数据类型(genbank, fasta/fastq),数据上传站点 2017/11/12 第二版:如何利用esearch, efecth快速获取SRR序列号 生物数据库 目前绝大部分数据由NCBI, EMBL-EBI, DDBJ三大机构托管,可划分为五类: (表格数据来源于INSDC) Data typeDDBJEMBL-EBINCBIN
biostar handbook: 第三周笔记汇总+第四周任务布置
就目前来看,这一周主要以补交上次作业为主,所以上一周的总结增加了很多新内容。但是关于本体论和富集分析,大家还是有点小困难。当然我自己交东西也慢了很多,因为时间也总是不太够,完全靠周末挤出来。 第三周笔记汇总如下 Biostar学习笔记(3)Gene set analysis related topics.本体论biostarhandboo(四)|本体论和功能分析基因本体论 第四周作业发布
biostar handbook: 第二周笔记汇总+第三周任务布置
第二周已经结束了,我不确定大家对Linux到底了解到了什么程度,但是我觉得再给一周时间让初学者去熟悉Linux肯定是必要的。于是这一周的任务不会太难,只需要让大家去理解本体论(ontology)。 笔记汇总 这周有一些小伙伴开始遭遇人生抉择,有一些则是要出差赶路,所以上交作业不算太多。可能大家对自己的要求有点高了,其实我一直强调的是笔记的不断迭代,只要你写了一点内容就可以发出来,后来不断修改,

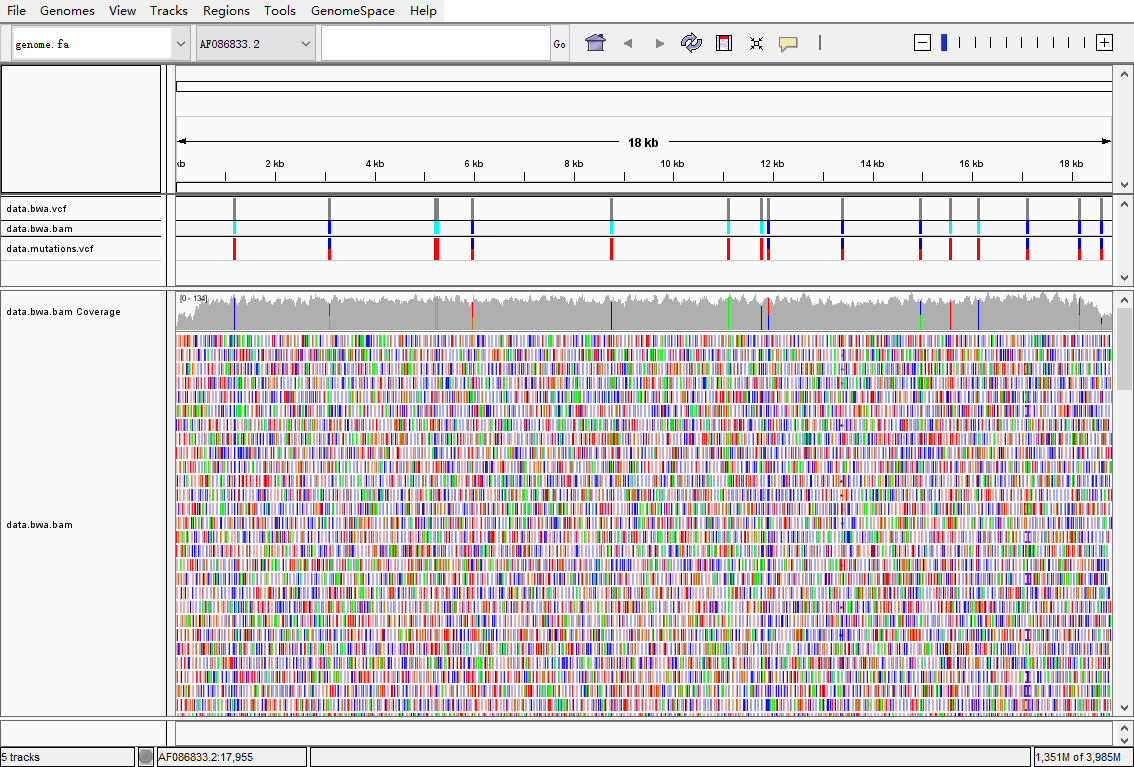
biostar handbook(十)|如何进行变异检测
变异检测流程 什么是基因组变异 基因组变异是一个定义比较模糊的概念. 所谓的变异是相对于一个完美的“参考基因组”而言。但是其实完美的“参考基因组”并不存在,因为我们只是选择某一个物种里的其中似乎比较正常的个体进行测序组装,然后基于它进行后续的研究。简单的说,参考和变异是相对而言,变异也可能完全正常。 常见的基因组变异一般可以归为如下几类: SNP, 单核苷酸多态性, 一
biostar handbook|如何模拟NGS测序结果
如何用软件模拟NGS数据 为了评价一个工具的性能,通常我们都需要先模拟一批数据。这样相当于有了参考答案,才能检查工具的实际表现情况。因此对于我们而言,面对一个新的功能,可以先用模拟的数据测试下不同工具的优缺点。有如下几个工具值得推荐一下: 'wgsim/dwgsim': 从全基因组中获取测序reads'msbar': EMBOSS其中一个工具,能够从单个序列中模拟随机突变'biosed': E

biostar handbook(一)分析环境和数据可重复
biostarhandbook(一)分析环境和数据可重复 2017/10/18/11:00第一版本笔记,主要更新了生物信息要用那些基本技能和电脑配置简单说明,以及如何在虚拟机器上安装bioconda 2017/10/18/12:30第二版笔记,增加虚拟机配置 2017/10/18/13:05 第三版笔记,增加文件结构 2017/10/19/10:30 第四版笔记, 增加如何用xshell连接虚
linux质控命令,Biostar_handbook||Charpter_6789_数据的格式_获取_质控_seqkit
Charpter6:数据的格式Data Formats 常用数据库 NCBI EBI Uniprot Phytozome Ensemble plants TAIR PlantGDB PlantTFDB 植物转录因子数据库 这些是我目前常用的一些数据库,可以看出来我是做植物的。其实植物领域还有许多重要的数据库,更不必说动物的了,各个物种都能拿出来做个数据库。数据这么多,如何更加高效的从数据库中实现