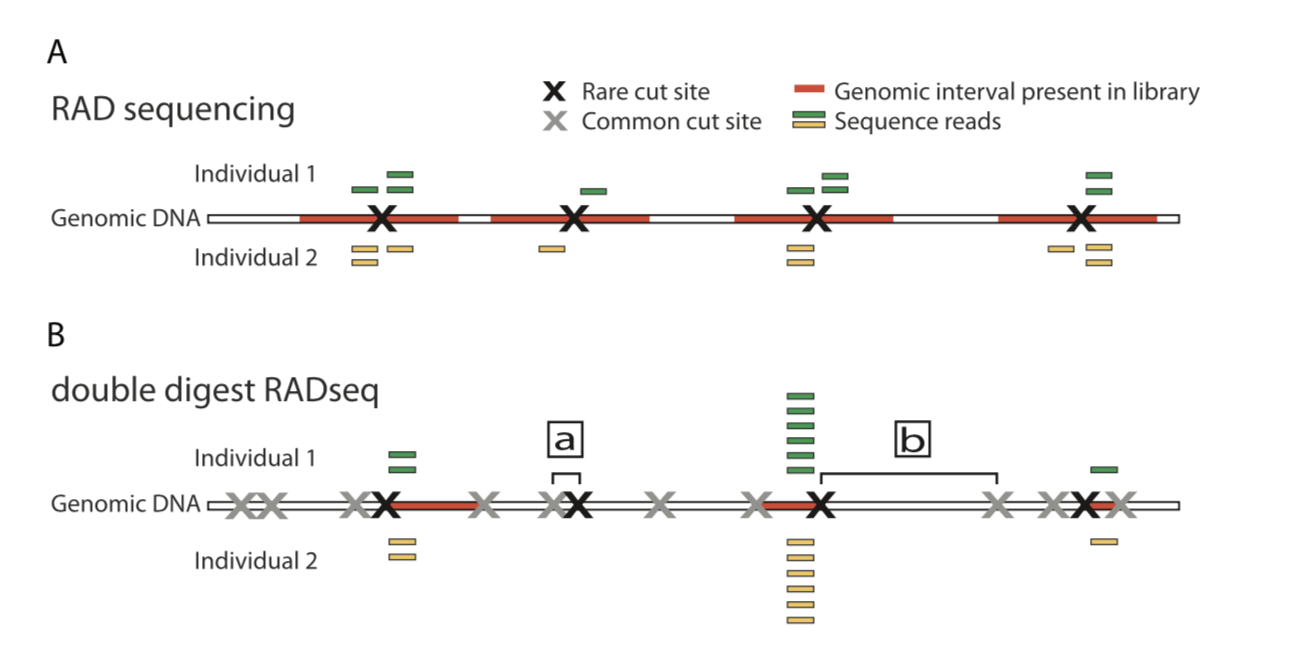
本文主要是介绍重测序(WGS)-群体遗传,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
进化论
- 达尔文进化论
- 生物物种存在广泛变异
- 自然选择,适者生存和繁殖,不适者淘汰
- 生命是单起源的
- 中性学说
- 大多数突变是“中性”的
- 进化主要是中性突变进行随机的遗传漂变
什么是群体?
概念
遗传学上的群体,与生态学上的种群,是一个概念。 种群 population:指同一时间生活在一定自然区域内,同种生物的所有个体。种群中的个体并不是机械地集合在一起,而是彼此可以交配,并通过繁殖将各自的基因传给可育后代。
种群有哪些特征?
空间特征 任何种群都要占据一定的分布区
数量特征
种群密度: 即单位面积或单位空间内的个体数目
生物量:即单位面积或单位空间所有个体的
遗传特征 形态、生理特征都有差异,本质在于遗传差异在环境影响下的表现
种群是物种存在和进化的基本单位

演化的基础
 自然选择 (natural selection):一个群体内遗传上不同的个体或基因型的有差别的增殖。有差别的增殖是由个体间如死亡率、能育性、生殖力、交配成功和后代生活力等因子方面的差异所造成的。自然选择被看成是进化的主要驱动力量 。(性选择)
自然选择 (natural selection):一个群体内遗传上不同的个体或基因型的有差别的增殖。有差别的增殖是由个体间如死亡率、能育性、生殖力、交配成功和后代生活力等因子方面的差异所造成的。自然选择被看成是进化的主要驱动力量 。(性选择)
遗传漂变 (genetic drift):是指当一个种群中的生物个体的数量较少时,下一代的个体容易因为有的个体没有产生后代,或是有的等位基因没有传给后代,而和上一代有不同的等位基因频率。(自然发生的随机事件,不可避免)
迁移(migration):当族群之间并未受到地理或是文化上的阻碍时,基因变异会经由一些个体的迁移,使基因在不同族群间扩散,这样的情形称为基因流。
群体选择
- 正选择
当一个群体中出现能够提高个体生存力或育性的突变时,具有该基因的个体将比其它个体留下更多的子代,而突变基因最终在整个群体中扩散。
有利突变产生 - 受到选择 - 选择性清除
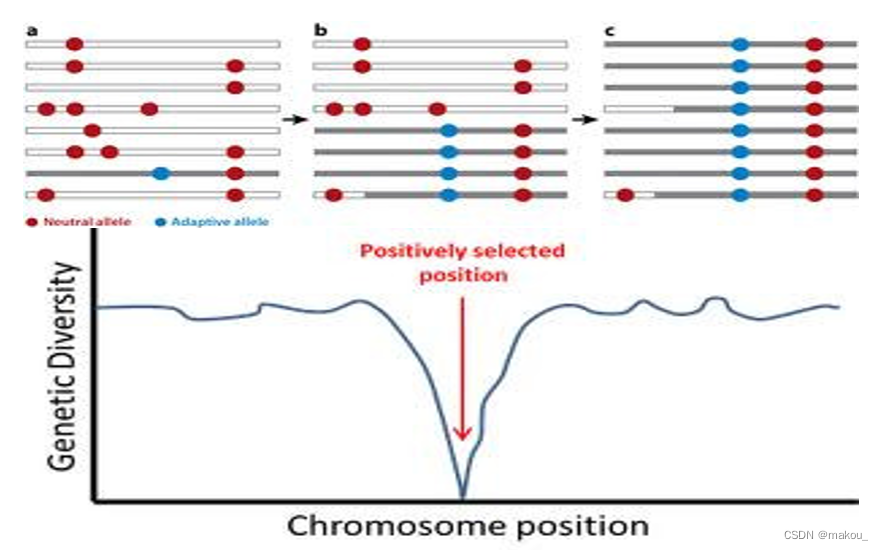
正选择的特征及检验方法
- 受选择位点的遗传多态性降低
- 引起选择性清除( Selective Sweep )
– 当某一特定位点上的基因或等位基因受到自然选择的强烈影响,使得该位点上的有利等位基因的频率显著增加,而其他等位基因被清除或减少。结果是,该位点两侧的序列多态性(即基因的变异)会因为连带效应而保持很低水平。 - 引起选择搭载效应(hitchhiking effort)
– 在选择性清除中,有时候与正选择位点相邻的连锁位点(非直接受选择影响的位点)的频率也会上升。这是由于选择性清除的作用,使得正选择位点附近的连锁位点也受到影响,导致它们的频率发生变化。这个现象被称为选择搭载效应。 - 引起连锁不平衡的增加-选择搭载效应可能导致连锁位点之间的连锁不平衡增加。连锁不平衡是指两个或多个基因座之间的关联程度,其变化可能是由于选择性清除引起的。增加的连锁不平衡意味着这些位点之间的关联性增强,因为它们一起经历了自然选择。
负选择
指群体中出现有害突变时,携带该基因的个体会因为生存力或育性降低而从群体中淘汰。
- 有害突变 - 淘汰 - 多态性降低
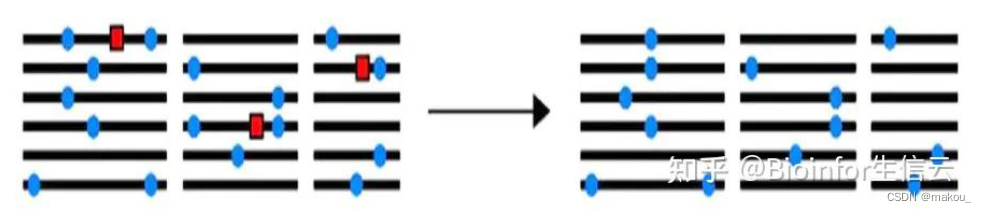
平衡选择
指多个等位基因在一个群体的基因库中以高于遗传漂变预期的频率被保留。
如杂合子优势
如稀有突变有更高的适应性
群体基因组学研究什么?
- 通过分析DNA序列多态数据,推测曾经作用于基因组的各种力量,进而探讨生物演化的过程。
- 研究内容主要包括群体结构和数量变化、群体突变、自然选择、遗传漂变等,以及这些与基因组中的遗传变异关系。
- 是从全基因组层面系统揭示群体遗传差异的学科,是群体遗传学的深入和延伸。
群体基因组学的研究手段
个体基因组多态性分析
——重测序分析(Re-sequencing)
——全基因组关联分析(Whole Gnome Association Analysis)
个体基因组更全面的差异分析
——泛基因组学分析(pan genome)
重测序(re-sequencing)——利用群体个体的基因组多态性分析进行群体遗传学研究
全基因组重测序是对有已知基因组序列的物种:
针对不同特征的个体,进行基因组测序;
通过与参考基因组进行序列比对,检测到大量变异信息,包括单核苷酸多态性(SNP)、插入缺失(InDel)、结构变异(SV)和拷贝数变异(CNV)等;
基于检测到的遗传变异,进一步研究动植物的物种特性、群体进化问题、定位目标性状基因位点。
基于参考基因组的群体水平重测序分析——变异的发掘和深度后续分析

基于群体间分歧度检验-Fst
Fst(Fixation index)是用于衡量群体间遗传变异的一种统计指标。它常被用于遗传学和种群遗传学的研究中,特别是在描述不同群体或亚群体之间的基因频率差异时。
Fst的值范围是从0到1,表示在总遗传变异中由于群体间的差异而产生的比例。不同的Fst值有不同的意义:
- Fst = 0: 表示群体之间没有基因频率的差异,即群体间基因池完全相同。
- Fst 接近 0: 表示群体之间的基因频率差异很小,即存在一些差异,但这些差异相对较小。
- Fst 接近 1: 表示群体之间的基因频率差异很大,即群体间基因池差异很显著。
Fst的解释还可以按照以下范围进行更详细的分类:
- 低(Low): 0 < Fst < 0.05
- 中等(Moderate): 0.05 ≤ Fst < 0.15
- 高(High): Fst ≥ 0.15
Fst值的解释还需要结合具体的研究背景和问题来看。在一些研究中,即使Fst值较小,也可能有重要的生物学意义,尤其是当研究关注特定基因座或与适应性有关的基因时。反之,Fst值较大可能意味着群体间存在较大的分化,可能是由于地理障碍、生态因素或人为选择等原因。
总体而言,Fst是评估群体间遗传结构和基因流的有力工具,能够提供关于群体演化历史和遗传多样性的重要信息。
Fst出现负值
在理论上,Fst的取值范围应该是0到1之间,因为它表示遗传变异中由于群体间差异导致的比例。然而,Fst在实际计算中偶尔可能呈现负值。这通常是由于计算过程中的数值问题、样本大小差异或者遗传标记的性质等原因引起的。
主要的原因之一是采用的遗传标记的多态性较低,导致了一些计算中的困难。当某个位点上的变异性非常有限时,Fst的计算可能会受到限制。此外,当样本容量较小时,由于统计噪声的影响,Fst的估计也可能变得不稳定。
解决这个问题的方法通常包括:
- 检查数据: 检查数据中是否存在错误或异常值,确保数据质量。
- 使用更多的遗传标记: 增加使用的遗传标记数量,以提高Fst的稳定性和准确性。
- 考虑样本大小: 在计算Fst时,确保各个群体的样本大小相对均衡,以减少样本大小差异可能导致的影响。
- 使用不同的计算方法: 有时候,使用不同的Fst计算方法或调整计算的参数可能有助于解决负值的问题。
虽然Fst的负值可能是数值计算问题的结果,但在解释数据时,研究人员通常会将其视为0,因为Fst不能为负。在实际应用中,对数据的审查和验证是非常重要的,以确保Fst的结果是可靠和合理的。
权重Fst(Weight Fst)和平均Fst
"权重 Fst" 和 "平均 Fst" 都是衡量群体间遗传差异的指标,但它们在计算方式和解释上有一些不同。
- 平均 Fst: 这是一种简单的平均值,计算所有群体间的 Fst 并取平均。它不考虑各群体的大小或重要性,对每个群体的贡献都一视同仁。计算公式为:

- 权重 Fst: 这是对平均 Fst 进行改进,考虑了群体大小或其他重要性因素。不同群体的贡献被赋予不同的权重,通常是根据群体的大小。这是因为在样本较小的群体中,由于随机性的影响,估计的 Fst 可能会更不稳定,因此可以给予较大的群体更高的权重。计算公式可以表示为:

在实际应用中,选择使用平均 Fst 还是权重 Fst 取决于研究的具体情况。如果群体大小差异较大,且希望更加重视较大群体的贡献,那么可以考虑使用权重 Fst。否则,平均 Fst 在某些情况下可能更为简便,特别是当群体大小相对均衡时。
多样性参数-Pi(π)
核苷酸多样性是一个反映基因组内或基因座上的遗传多样性水平的指标。在分子生物学和遗传学的研究中,通常用核苷酸多样性来描述DNA序列的变异程度。核苷酸多样性的取值范围是从0到1,具体的意义如下:
- 核苷酸多样性为 0: 表示在该基因座或 DNA 区域上,所有个体的 DNA 序列都是一样的,没有任何变异。这可能表明这个基因座在群体中高度保守,没有发生多态性。
- 核苷酸多样性为 1: 表示在该基因座或 DNA 区域上,所有个体的 DNA 序列都是完全不同的,每个个体都有独特的 DNA 序列。这意味着这个基因座在群体中非常多态,存在大量的变异。
- 中间值的核苷酸多样性: 在 0 和 1 之间的值表示中等水平的多态性。这表示在群体中有一些变异,但并非每个个体的 DNA 序列都是独特的。
核苷酸多样性的计算通常涉及到统计学上的一些指标,比如核苷酸多样性指数(nucleotide diversity index)或π值。这些指标可以通过比较同一基因座或 DNA 区域上不同个体的 DNA 序列,来估计遗传多样性的水平。简单来说就是所取样本序列中,任意两序列之间每个位点上核苷酸差异的平均数。
核苷酸多样性的理解对于遗传变异、种群遗传学、进化生物学等领域的研究非常重要,因为它提供了有关群体内基因多样性的信息,有助于了解物种的演化历史、适应性和群体结构。
Pi == 0.004
核苷酸多样性为0.004表示在考察的基因座或 DNA 区域上,个体间的 DNA 序列变异程度相对较低。具体来说:
- 低水平的遗传多样性: 核苷酸多样性为0.004表明群体中的 DNA 序列在这个基因座上变异程度较小。这样的多态性水平通常被认为是相对较低的。
- 较为保守的基因座: 0.004的核苷酸多样性值相对较小,说明该基因座在群体中相对保守,即DNA序列的变异相对较少。
- 缺乏显著的遗传变异: 核苷酸多样性为0.004意味着个体之间的DNA序列差异相对较小,基因座在群体中可能缺乏显著的遗传变异。
这样的结果可能有多种解释,包括该基因座可能在群体中比较保守,或者该群体可能经历了一定程度的遗传漂变或选择,导致基因座上的变异较为有限。
总体而言,核苷酸多样性为0.004的情况表明在该基因座上,群体中的个体之间的DNA序列变异相对较小。这样的信息对于理解群体的遗传结构和基因座的演化特征具有重要意义。
群体多态性参数θ
遗传多态性:在一个群体中 ,存在两个或者多个有着相当高频率(通常大于1%)的等位基因时就称为遗传多态性。
Ne为有效群体大小,μ为每个位点的突变速率,群体多态性参数θ定义为:

θ常用的估算:
- 分离位点数目(θw)
分离位点是指相关基因在多序列比对中表现出多态性的位置。 - 核苷酸多样性 π(核苷酸水平杂合度)
样本群体中两两序列间每个位点上核苷酸差异的平均值
Tajima'D
Tajima's D 是一种中性进化的检验统计量,通常用于评估在一群物种或种群中,基因座是否经历了中性演化。这个统计量由日本遗传学家 Tajima 提出,主要基于两个遗传变异的度量:多态性(多态位点的数量)和平均杂合度(Heterozygosity)。Tajima's D 的计算公式如下:

其中:
- π 是多态性,表示两个随机选择的基因座在样本中不同的碱基对的比例。
- θ 是平均杂合度,表示两个随机选择的基因座在样本中具有不同碱基的概率。
- a 是一个校正因子,通常通过样本大小和多态性的期望值来估计。
Tajima's D 的理论期望值依赖于群体的演化历史,包括群体的大小、漂变率和迁移率。在中性演化的情况下,理论期望值为0。正的 Tajima's D 可能表明过去的人口缩减或选择的效应,而负的 Tajima's D 可能表明过去的人口扩张或整体中性演化。
在中性检验中,常见的情况是根据 Tajima's D 的显著性水平来判断一个基因座是否经历了中性演化。如果 Tajima's D 在统计上显著不同于0,那么可能表明该基因座经历了非中性演化,而不是简单的随机演化。
需要注意的是,Tajima's D 的解释需要谨慎,因为它的结果受到许多因素的影响,包括种群历史、选择压力和样本的大小。因此,在解释 Tajima's D 的结果时,通常需要将其与其他证据和背景信息结合起来。
Tajima'D取值范围
Tajima's D 的理论取值范围通常是从负无穷到正无穷。不同的取值反映了不同的演化历史和群体动力学。具体的解释如下:
- Tajima's D = 0: 在中性演化的情况下,Tajima's D 的期望值为0。因此,如果观察到 Tajima's D 等于0,可能表明该基因座经历了中性演化。
- Tajima's D > 0: 正的 Tajima's D 可能暗示了一些非中性演化的事件,如种群缩减、选择或其他非随机的遗传过程。这可能表明较多的低频变异。
- Tajima's D < 0: 负的 Tajima's D 可能表明过去的人口扩张或其他非中性演化的事件。这可能表明较多的高频变异。
Tajima'D值为正
Tajima's D 的理论取值范围通常是从负无穷到正无穷,如 Tajima's D = 2 是一个相对较大的正值。这样的结果可能需要谨慎解释,因为 Tajima's D 通常在中性演化的情况下期望值为 0。
一般而言,Tajima's D 为正可能意味着群体历史中发生了一些非典型的事件,比如:
- 种群收缩: 正的 Tajima's D 值可能是由于过去发生的种群收缩事件导致的。在种群收缩期间,由于随机漂变的作用,会导致较多的低频变异。
- 正选择: 正选择(positive selection)也可能导致 Tajima's D 值为正。正选择促使有益基因变异的快速传播,从而导致较多的低频变异。
- 其他非典型演化事件: 其他群体动力学的变化或非典型的演化事件也可能导致 Tajima's D 为正。
需要指出的是,具体解释 Tajima's D 的值需要根据研究背景和其他证据进行综合考虑。此外,样本大小的影响也需要注意,较小的样本可能导致 Tajima's D 的估计更加不稳定。
如果 Tajima's D 的值在统计上显著不同于0,这可能提示着与中性演化不符的群体历史或遗传过程。在这种情况下,进一步的调查和分析可能是必要的,以更好地理解所观察到的遗传多样性模式。
Tajima'D值为负
当 Tajima's D 为负时,这通常表明群体遗传多样性的模式与中性演化的预期值不同。负的 Tajima's D 可能由以下一些情况引起:
- 人口扩张: 一种常见的解释是过去发生了人口扩张。在人口扩张期间,由于新的变异被引入群体,会导致较多的高频变异,从而使 Tajima's D 变为负值。
- 正选择: 正选择(positive selection)也可能导致 Tajima's D 为负。正选择推动有益变异的迅速传播,导致较多的高频变异。
- 群体分化: 群体分化或亚群体之间的基因流减少也可能导致 Tajima's D 为负。在这种情况下,不同的亚群体可能累积了各自的高频变异。
- 漂变: 随机漂变的效应可能导致 Tajima's D 为负,尤其是在样本大小相对较小的情况下。
需要注意的是,负的 Tajima's D 值并不一定意味着负选择或人口扩张等特定事件,而可能是多种复杂因素的结果。因此,对 Tajima's D 的解释需要谨慎,并最好结合其他证据和背景信息。此外,负的 Tajima's D 也可能是由于样本中的一些特殊情况或方法ological artifacts 引起的,因此在解释结果时需要考虑数据质量和分析方法。
XP-CLR
XP-CLR(Cross Population Composite Likelihood Ratio)是一种用于检测群体间选择信号的计算方法。它主要应用于对不同人群或种群之间的基因型数据进行比较,以发现在进化过程中受到自然选择影响的基因区域。
XP-CLR 的主要思想是比较两个或多个群体之间的基因型频率差异,以识别可能受到选择压力的基因区域。该方法通过计算一个综合的似然比(composite likelihood ratio)分数来实现这一目标。
XP-CLR 的计算过程涉及以下步骤:
- 建立模型: 首先,对于每个基因座,建立一个模型来描述不同群体之间的基因型频率差异。这通常包括考虑突变率、基因型频率和迁移率等因素。
- 计算似然比: 使用群体之间的基因型数据,计算在选择存在和不存在的两种假设下的似然比。这反映了数据在两个模型之间的拟合优度差异。
- 归一化: 对似然比进行归一化,以控制数据集的大小和复杂性。
- 标准化分数: 将得到的归一化似然比转化为标准化分数,用于识别可能受到选择压力的基因区域。
XP-CLR 方法的输出通常是一系列标准化的分数,用于表示基因组上的不同区域是否受到选择的影响。这些分数没有明确的固定范围,因为它们是从似然比得到的,并且具体的范围可能受到分析中使用的数据集和方法的影响。在一般情况下,XP-CLR 分数越高,表示在相应的基因区域内,群体间的基因型频率差异越显著,可能受到了选择的影响。分数的具体解释通常需要与研究的具体背景和目标相结合。在实际应用中,研究者通常会使用一些阈值或统计方法来识别显著的选择信号。这些阈值的确定可能涉及到模拟研究、对照组的建立和比较,以及对群体间遗传结构的了解。因此,XP-CLR 分数的具体取值范围的解释可能需要参考具体研究或文献中的定义和说明。XP-CLR 方法在发现群体间选择信号方面具有一定的优势,尤其是当考虑到多个群体时。它在人类基因组研究和其他物种的遗传学研究中得到了广泛应用,有助于理解基因在不同群体间的遗传变异和选择过程。
参考资料:
作者:李一城 链接:https://zhuanlan.zhihu.com/p/667488075
作者:Bioinfor生信云 链接:https://zhuanlan.zhihu.com/p/590071862
这篇关于重测序(WGS)-群体遗传的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!