本文主要是介绍Paper - DeepMSA2: Improving deep learning protein monomer and complex structure prediction,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
欢迎关注我的CSDN:https://spike.blog.csdn.net/
本文地址:https://spike.blog.csdn.net/article/details/135520805
DeepMSA2 是用于构建高质量的蛋白质单体和复合体多序列比对(MSA)的流程,利用了迭代的序列搜索和隐马尔可夫模型算法,从多个基因组和元基因组数据库中提取了大量的同源序列。DeepMSA2 的主要优势在于平衡的序列搜索和有效的模型选择,以及利用海量元基因组数据库的能力,这些结果表明通过改进 MSA 的构建,可以为深度学习蛋白质结构预测提供一个新的途径,也证明了优化深度学习方法的输入信息,与设计预测器本身一样重要。
Paper:
- DeepMSA2: Improving deep learning protein monomer and complex structure prediction using DeepMSA2 with huge metagenomics data - Nature Methods 2024.1.3
- DMFold: A deep learning platform for protein complex structure and function predictions based on DeepMSA2
源码:
- DMFold:https://zhanggroup.org/DMFold
- DeepMSA:https://zhanggroup.org/DeepMSA/download/
DMFold:
DMFold (also known as DMFold-Multimer) is a deep learning-based approach to protein complex structure and function prediction built on deep multiple sequence alignments (MSAs).
DMFold(也称为 DMFold-Multimer)是一种基于深度学习的蛋白质复合物结构和功能预测方法,建立在深度多序列比对(MSAs)的基础上。
The core of the pipeline is the integration of DeepMSA2 with the modified structure module of AlphaFold2.
该流程的核心是将 DeepMSA2 与修改后的 AlphaFold2 结构模块集成在一起。
Starting from a set of query sequences, DMFold first creates deep monomeric MSAs using an iterative search procedure through multiple whole-genome (Uniclust30 and UniRef90) and metagenome (Metaclust, BFD, Mgnify, TaraDB, MetaSourceDB and JGIclust) databases, where multimeric MSAs are then constructed by pairing the monomeric MSAs based on species annotations.
从一组查询序列开始,DMFold 首先使用多个全基因组(Uniclust30和UniRef90)和宏基因组(Metaclust,BFD,Mgnify,TaraDB,MetaSourceDB和JGIclust)数据库的迭代搜索过程,创建深度单体 MSAs,然后根据物种注释将单体 MSAs 配对构建多体 MSAs。
Next, complex structure models are predicted by feeding the multimetic MSAs into the structural modules of AlphaFold2-Multimer, where funtional annotations, including Gene Ontology, Enzyme Commission and Ligand Binding Sites, are generated by COFACTOR2 and US-align based on the top DMFold structure models.
接下来,通过将多体 MSAs 输入到 AlphaFold2-Multimer 的结构模块中,预测复合物结构模型,然后根据 DMFold 的最优结构模型,由 COFACTOR2 和 US-align 生成功能注释,包括基因本体、酶分类和配体结合位点。
DMFold participated (as “Zheng”) in CASP15 and ranked as the No. 1 method for protein-protein complex structure prediction, with accuracy significantly higher than the state-of-the-art AlphaFold2 program (i.e., “NBIS-AF2-multimer” in CASP15).
DMFold 以 “Zheng” 为名,参加 CASP15,并在蛋白质-蛋白质复合物结构预测方面排名第一,其准确度显著高于最先进的 AlphaFold2 程序(即CASP15 中的 “NBIS-AF2-multimer”)。
Although DMFold focuses on multi-chain protein complexes, it also accepts single-chain monomer sequences (DMFold-Monomer pipeline). The server is freely accessible to all users, including commercial ones.
尽管 DMFold 专注于多链蛋白质复合物,但是也接受单链单体序列(DMFold-Monomer流程)。该服务器对所有用户(包括商业用户)免费开放。
DeepMSA:
DeepMSA2 standalone package is a program for deep multiple sequence alignment generation for both monomer and multimer proteins.
DeepMSA2独立包是一个用于生成单体和多体蛋白质的深度多序列比对的程序。
Please report bugs and questions at Zhang Lab Service System Discussion Board. The DeepMSA2 package is free for academic and non-profit researchers.
请在 Zhang 实验室服务系统讨论板上报告错误和问题。DeepMSA2包对学术和非营利研究者免费。
工程下载:
- https://zhanggroup.org/DeepMSA/download/download_DeepMSA2.cgi?ID=112358=DeepMSA2-2.0.zip,源码约 3.8G
- https://zhanggroup.org/DMFold/download/download_DMFold.cgi?ID=235813=DMFold-1.0.zip,源码约 6.5G
补充数据 JGI (JGIclust30) list,参考源码的Download_lib.py,即:
DB.fasta.aa
DB.fasta.ab
DB.fasta.ac
DB.fasta.ad
1. DeepMSA2 Monomer

dMSA: The dMSA algorithm used in DeepMSA2 is modified from our previous MSA generation tool, DeepMSA.
DeepMSA2中使用的dMSA算法是从我们之前的MSA生成工具DeepMSA修改而来的。
qMSA(quadrupole MSA): The qMSA is composed of four stages that perform HHblits (v.2), Jackhmmer, HHblits (v.3) and HMMsearch searches against Uniref30, Uniref90, BFD and Mgnify databases, respectively.
qMSA由四个阶段组成,分别对Uniref30, Uniref90, BFD和Mgnify数据库进行HHblits (v.2), Jackhmmer, HHblits (v.3)和HMMsearch搜索。
mMSA: the qMSA stage 3 alignment is used as a probe by HMMsearch using parameters ‘-E 10 --incE 1e-3’ to search through a metagenomics database combining JGIclust, TaraDB and MetaSourceDB, with the resulting sequence hits converted to a raw sequence database.
在mMSA中,qMSA第3阶段的比对结果被用作HMMsearch的探针,使用参数‘-E 10 --incE 1e-3’在一个由JGIclust, TaraDB和MetaSourceDB组合的宏基因组数据库中进行搜索,将搜索到的序列命中结果转换为原始序列数据库。
JGI 的 clust 数据是单独提供,参考 DeepMSA2/Download_lib.py:
- JGIclust collected from IMG/M, MetaSourceDB, TaraDB
######### download JGI (JGIclust, MetaSourceDB and TaraDB are included in here)
if not os.path.exists("JGIclust"):os.system("mkdir -p JGIclust")
os.chdir("JGIclust")os.system("wget -c https://zhanggroup.org/ftp/data/JGIclust30/list")
jgiclust_db_list=[]
jgifile=open('list','r')
lines=jgifile.readlines()
jgifile.close()
for line in lines:jgiclust_db_list.append(line.strip('\n'))for jgidb in jgiclust_db_list:print("download jgi db %s"%jgidb)os.system("wget -c https://zhanggroup.org/ftp/data/JGIclust30/%s.xz"%jgidb)print("decompress jgi db %s"%jgidb)os.system("xz -dvf %s.xz"%jgidb)print("download jgi ssi db %s"%jgidb)os.system("wget -c https://zhanggroup.org/ftp/data/JGIclust30/%s.ssi.xz"%jgidb)print("decompress jgi ssi db %s"%jgidb)os.system("xz -dvf %s.ssi.xz"%jgidb)
相关 MSA 库的规模,参考:

JGIclust30 的下载地址:https://zhanggroup.org/ftp/data/JGIclust30/

2. DeepMSA2 Multimer

MSA Pairing
MSA pairing. Two types of complexes are considered in DeepMSA2.
MSA配对。DeepMSA2考虑了两种类型的复合物。
For homomeric complexes in which all component chains are identical, all of the monomer MSAs are utilized and the multimeric MSAs are created by concatenating each of the monomer MSAs n times side-by-side, where n is the number of monomer chains.
对于同源复合物,所有的组分链都是相同的,所有的单体MSA都被利用,多聚体MSA是通过将每个单体MSA侧向连接n次而生成的,其中n是单体链的数量。
For heteromeric complexes, the top M MSAs are selected for each monomer chain so that M^N distinct paired MSAs can be created for the complex, where N is the number of distinct chains in the complex.
对于异源复合物,为每个单体链选择排名最高的M个MSA,以便为复合物创建M^N个不同的配对MSA,其中N是复合物中不同链的数量。
To avoid an impractically long MSA construction time, M is set as the maximal value to satisfy M^N ≤ 100.
为了避免MSA构建时间过长,M被设定为满足M^N≤100的最大值。
For example, for a complex containing three different protein chains (A2B2C1, N = 3), M will be set to 4 (4^3 ≤ 100)
例如,对于一个包含三种不同蛋白质链(A2B2C1,N = 3)的复合物,M将被设定为4(4^3 ≤ 100)
In other words, for each component chain in this complex, we select four top-ranked monomer MSAs and build paired MSAs for the complex with 64 different combinations of those monomer MSAs.
换句话说,对于这个复合物中的每个组分链,我们选择四个排名最高的单体MSA,并用这些单体MSA的64种不同组合构建配对MSA。
Normally, MN ranges from 50 to 100 for different kinds of heteromer complexes.
通常,对于不同种类的异源复合物,M^N的范围在50到100之间。
Sequence linking
For a given set of M……N paired monomeric MSAs, the sequences from the monomeric MSAs are concatenated into a multimeric MSA as follows (Supplementary Fig. 12b).
对于给定的M^N个配对的单体MSA,将单体MSA中的序列连接成一个多聚体MSA
First, the sequences in each monomeric MSA are grouped based on the UniProt annotated species.
首先,根据UniProt注释的物种,将每个单体MSA中的序列分组。
The sequences in each group are then ordered based on the sequence identity to the query sequence.
然后,根据序列与查询序列的相似度,对每个组中的序列进行排序。
To properly capture orthologous signals of interchain coevolution, the top sequences of different monomeric MSAs belonging to the same species group are linked together side-by-side to form a composite sequence in the multimeric MSA.
为了正确捕捉链间共进化的同源信号,将不同单体MSA中属于同一物种组的顶部序列侧向连接在一起,形成多聚体MSA中的一个复合序列。
In cases where one of the monomeric MSAs is missing for a specific species, which appear in more than one other chains, the component chain is padded with gaps in the composite sequence with other linked chains having that species.
在某些情况下,如果某个物种在一个或多个其他链中出现,但是缺少对应的单体MSA,那么在与其他有该物种的链连接的复合序列中,用空位填充该组分链。
Finally, the unlinked sequences in the monomeric MSAs are padded below the linked sequences.
最后,将单体MSA中未连接的序列填充在连接序列的下方。
This composite linking step is applied only to heteromeric complexes, as the MSAs for homomeric complexes are constructed by simply concatenating the same monomer MSA multiple times.
这个复合连接步骤只适用于异源复合物,因为同源复合物的MSA,是通过简单地将同一个单体MSA多次连接而构建的。
扩展的 Uniref 物种信息源码:
def parse_sequence_specie(msa_sequence_identifier: str, dbtype='uniprot'):"""Gets species from an msa sequence identifier.The sequence identifier has the format specified by_UNIPROT_TREMBL_ENTRY_NAME_PATTERN or _UNIPROT_SWISSPROT_ENTRY_NAME_PATTERN.An example of a sequence identifier: `tr|A0A146SKV9|A0A146SKV9_FUNHE`Args:msa_sequence_identifier: a sequence identifier.Returns:An `Identifiers` instance with species_id. Thesecan be empty in the case where no identifier was found."""if dbtype=='uniref':matches = re.search(UNIREF_PATTERN, msa_sequence_identifier.strip())else:matches = re.search(UNIPROT_PATTERN, msa_sequence_identifier.strip())species_id=''if matches:species_id=matches.group('SpeciesIdentifier')return species_iddef get_species(id: str):"""get species from id"""sequence_identifier, dbtype = extract_sequence_ids(id)#print(sequence_identifier,dbtype)if sequence_identifier is None:return ''else:#print(parse_sequence_specie(sequence_identifier,dbtype=dbtype))return parse_sequence_specie(sequence_identifier,dbtype=dbtype)
MSA selection
Of the MN concatenated MSAs formed from the MSA paring procedure, 25 top MSAs are returned from the DeepMSA2-Multimer pipeline based on the M-score
从MSA配对过程中形成的M^N个连接的MSA中,根据M-score选择25个最优的MSA,由DeepMSA2-Multimer流程返回。
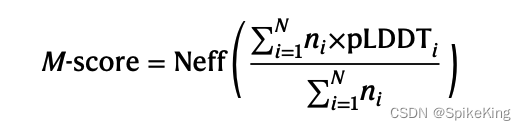
这篇关于Paper - DeepMSA2: Improving deep learning protein monomer and complex structure prediction的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!









