本文主要是介绍WGCNA如何挖掘潜在的共表达基因,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
欢迎关注微信公众号《生信修炼手册》!
共表达基因指的是表达量具有协同变化趋势的基因集合,通常认为这些基因参与相同的生物学过程,比如参与同一个代谢通路,正是由于功能上的协同作用,导致表达量呈现出高度相关性。
在WGCNA中,对传统的相关系数进行乘方运算,用最终得到的值来表征基因间的相关性。在计算出这样的相关性统计量值之后,如何确定哪些基因是共表达的呢?
WGCNA的做法是聚类分析,聚类分析属于一种非监督的机器学习算法,通过聚类树,可以观察到哪些基因在聚类树中属于同一分支,属于同一分支的基因可以归为一类。实际操作中,考虑到基因数目较多等情况,肯定需要算法来自动化的进行分类,WGCNA采用的是dynamicTreeCut这个R包。
对于聚类算法而言,需要输入基因间的距离矩阵,首先就需要将基因间的邻接矩阵转换为距离矩阵,对相关系数进行乘方运算,可以计算出邻接矩阵,但是这个值本质上反映的是基因间的相似度,并不是距离。在计算距离矩阵时,WGCNA采用了TOM这种统计量,该统计量可以表征网络中节点的相似性,计算公式如下
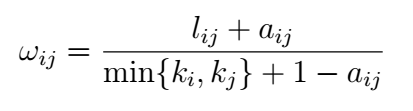
对于两个基因i和j而言,a表示两个基因邻接矩阵中对应的值,就是相关系数的乘方,K代表的每个基因的连接度, 公式如下
这篇关于WGCNA如何挖掘潜在的共表达基因的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!








