ncbi专题

NCBI-get-GCFIDs_fast.py
import requestsimport osimport redef download_genome_first(gcf_id):# 构建FTP下载路径base_url = "https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/"# 提取GCF号的数字部分并按三位分割parts = gcf_id.split('_')[1] # 提取数字部分path_

生信软件21 - 多线程拆分NCBI-SRA文件工具pfastq-dump
在使用NCBI 工具fastq-dump拆分SRA文件时,拆分速度慢, fastq-dump拆分参数说明: –split-spot: 将双端测序分为两份,存放在同一个文件中–split-files: 将双端测序分为两份,存放在不同的文件,但是对于一方有而一方没有的reads直接丢弃–split-3 : 将双端测序分为两份,存放在不同的文件,但是对于一方有而一方没有的reads会单独放在一个文件
sra数据下载linux,NCBI-SRA数据的下载方法
SRA 数据库: 为Sequence Read Archive 的缩写。主要存储高通量测序的原始数据,来自四个测序平台,分别为:Roche_LS454,Illumina,ABI_SOLID和HELICOS。从事生物信息分析的老师和同学一般都会接触SRA数据,下载SRA数据的方法也有很多,这里来简单总结一下。 一、SRA Tookit下载 选择需要的SRA Tookit 版本进行下载,下载后直接
NCBI下载SRA数据
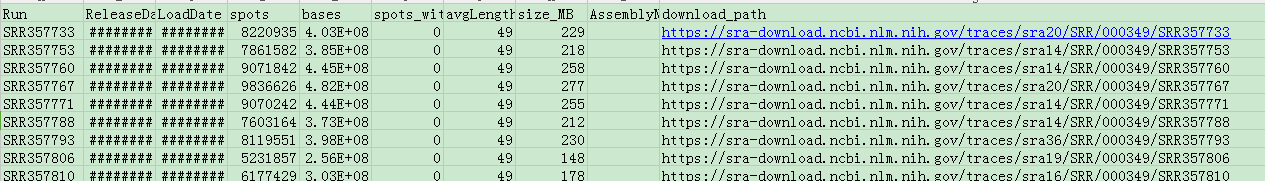
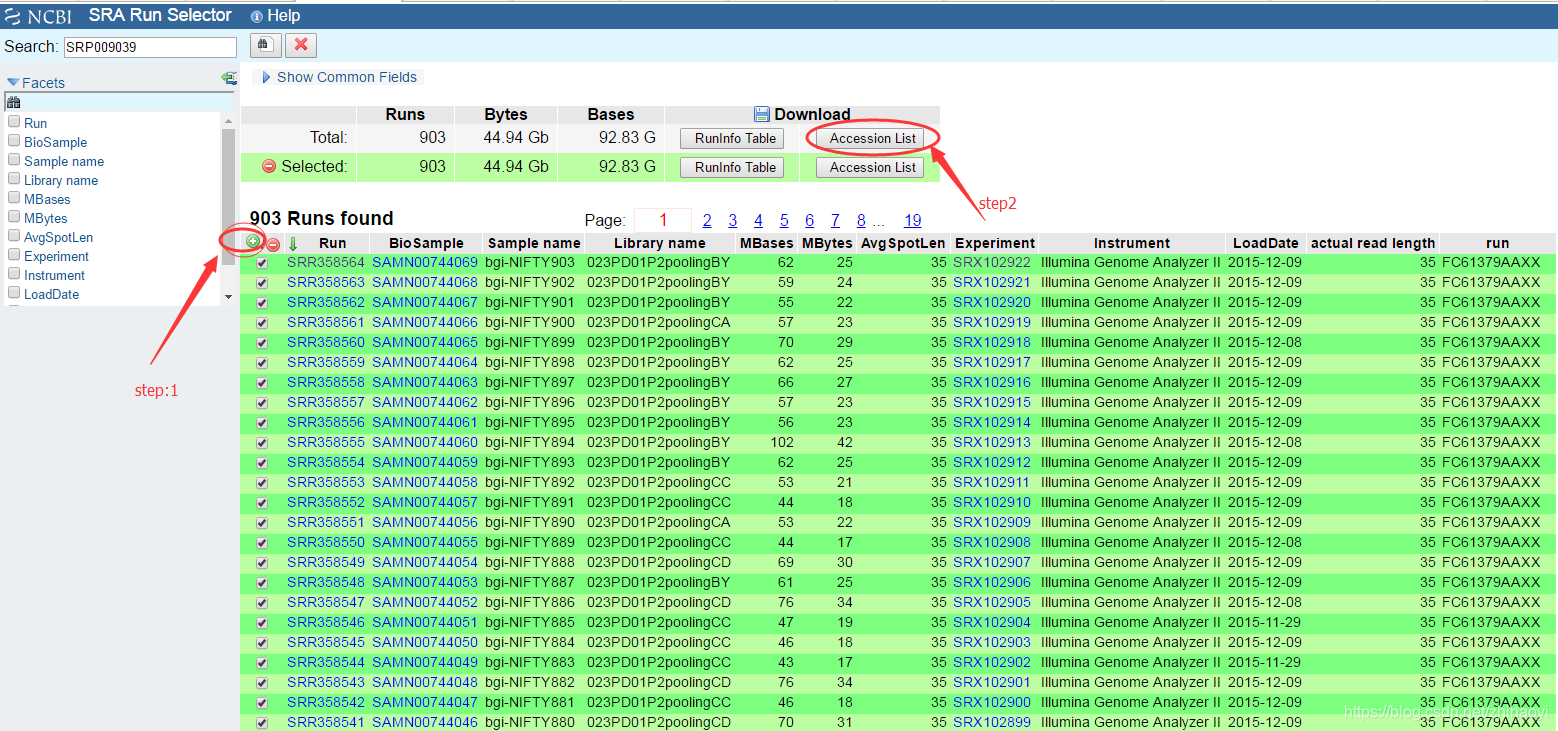
从NCBI下载数据本来是一件很简单的事情,但是今天碰到几个坑: 1、paper里没有提供SRA数据号、也没有提供路径; 2、不知道文件在ftp的地址,不能直接用wget下载 所以通过在NCBI官网,直接在SRA搜索栏里: 输入paper的title关键词NIFTY BGI 搜索结果: 选一个文件点击进去 进去之后,再点击SRP 然后: 出现如下内容: 然后选择所有SRR
NCBI下载SRA数据(转)
从NCBI下载数据本来是一件很简单的事情,但是今天碰到几个坑: 1、paper里没有提供SRA数据号、也没有提供路径; 2、不知道文件在ftp的地址,不能直接用wget下载 所以通过在NCBI官网,直接在SRA搜索栏里: 输入paper的title关键词NIFTY BGI 搜索结果: 选一个文件点击进去 进去之后,再点击SRP 然后: 出现如下内容: 然后选择所有SRR文件:

如何在NCBI实现大批量数据的一一对应
有时我们手头会有一批数据,或者是只有大量的某些id。比方说:accession number、gi、geneid、symbol、go、unigene、pubmed、taxid等等。事实大部分数据库都会有提供一些专门的文件或工具来实现这些数据间大批量的一一对应。 先来讲讲NCBI的。 用FTP登陆ftp.ncbi.nih.gov(windows下可以直接打开或是用迅雷/Flastget等下载工具

使用Python ete3包快速大批量地寻找物种的NCBI Taxonomy分类的完整信息(物种名和Taxid可进行相互转换)
ete全称为Environment for Tree Exploration,直译就是树探索环境,此工具可以直接在终端输入 pip install ete3 进行安装即可。ete包主要功能与构建系统发生树有关,若是有相关需求可以查看其介绍文档,地址:The ETE tutorial。我主要使用到了其中的分类工具,即处理NCBI 的Taxonomy数据库的工具。此工具用于物种信息和分类号的转换十分简

Biopython从NCBI搜索和取回数据库记录
Entrez模块 Entrez提供了链向在NCBI服务器的esearch和efetch工具的连接 列出Entrez模块的方法和属性 #!/usr/bin/env python# -*- coding: utf-8 -*-__author__ = 'sunchengquan'__mail__ = '1641562360@qq.com'from Bio import Entrezs

ncbi-genome-download批量下载基因组数据
1. ncbi-genome-download 的下载和安装 ncbi-genome-download 是一个可以直接从NCBI上批量下载序列的软件,支持下载多种格式 利用 conda 对其直接安装 参考 #创建环境conda create -n ncbi_genome_download#激活环境conda activate ncbi_genome_download# 安装cond
从NCBI测序数据下载,相关软件安装,到FastQC使用
1. 从ncbi下载测序数据 SRA链接:https://www.ncbi.nlm.nih.gov/sra 检索所需的项目,这里以Whole genome sequencing of ExPECs (SRR24129389)为例。 wget -c -t 0 -O ./SRR24129389.sra https://sra-pub-run-odp.s3.amazonaws.com/sra/S

Python爬虫获取geneID对应的NCBI注释
在海量的组学数据中,我们经常需要根据已有的差异表达基因找到对应的注释信息。那么针对一系列基因ID批量获取其注释无疑能够大大简化后继的分析,提高科研效率。本次来分享使用python爬虫完成NCBI基因注释的方法。 Sample input: 输入文件如下,是一列geneID。 待获取的信息来源于NCBI-geneID页中Description项,也就是下图中红色方框项: Sample
中美数据实现实时共享:首批中国自主收录的新冠病毒基因组序列在CNBC/NGDC和NCBI同时发布...
疫情下的大数据力量! 世卫呼吁各国提高新型冠状病毒数据分享,点“这里”查看。 大数据产业创新服务媒体 ——聚焦数据 · 改变商业 据中国科学院北京基因组研究所的官网显示,国家生物信息中心(CNCB)/国家基因组科学数据中心(NGDC)首批自主收录的5株2019新型冠状病毒基因组序列与美国NCBI核酸数据库GenBank数据实现了同步与共享。 CNCB/NGDC建立的2019新型冠状病毒信
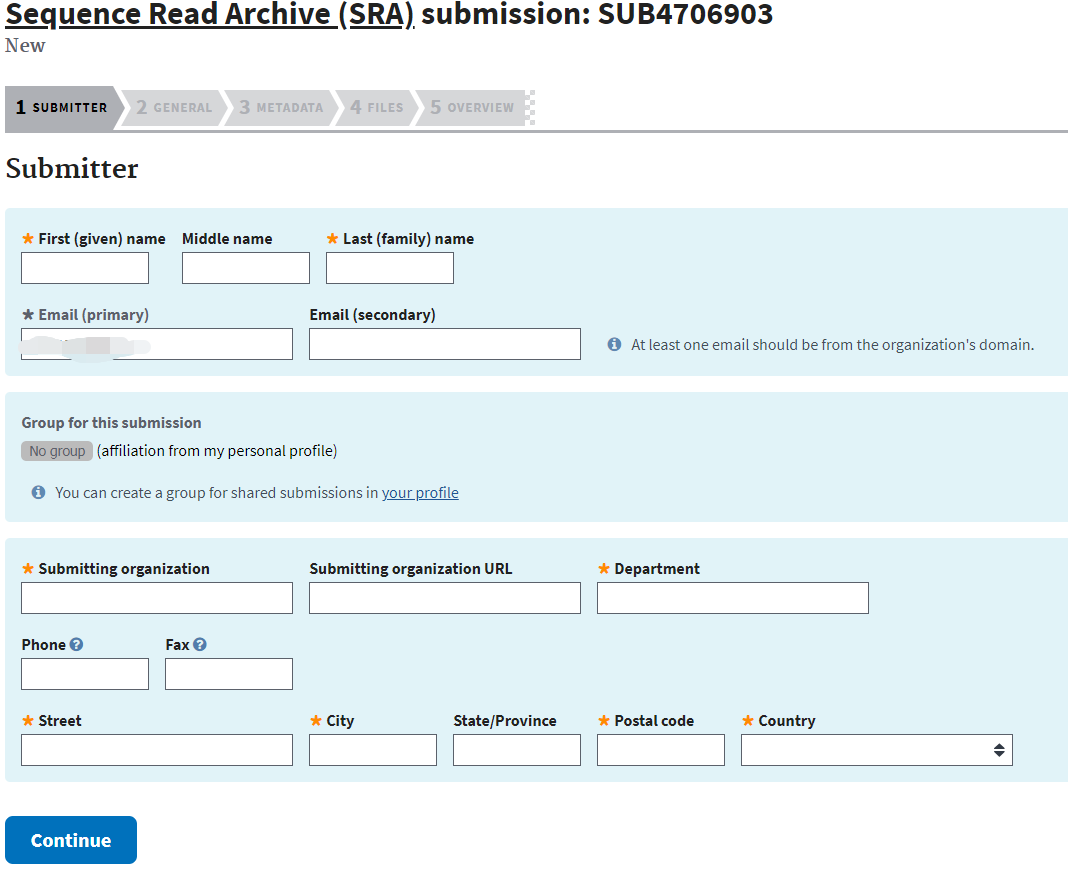
如何向NCBI的SRA上传数据
首先访问SRA的提交界面,https://submit.ncbi.nlm.nih.gov/subs/sra/,在没有登陆NCBI时,网页内容如下所示 未登录时 点击Log in, 会进入一个新的网页用于登陆或者创建新的NCBI账户。 登陆NCBI 在创建完账号或者登陆之后,返回之前的SRA上传页面,界面就变成了如下。 登陆后
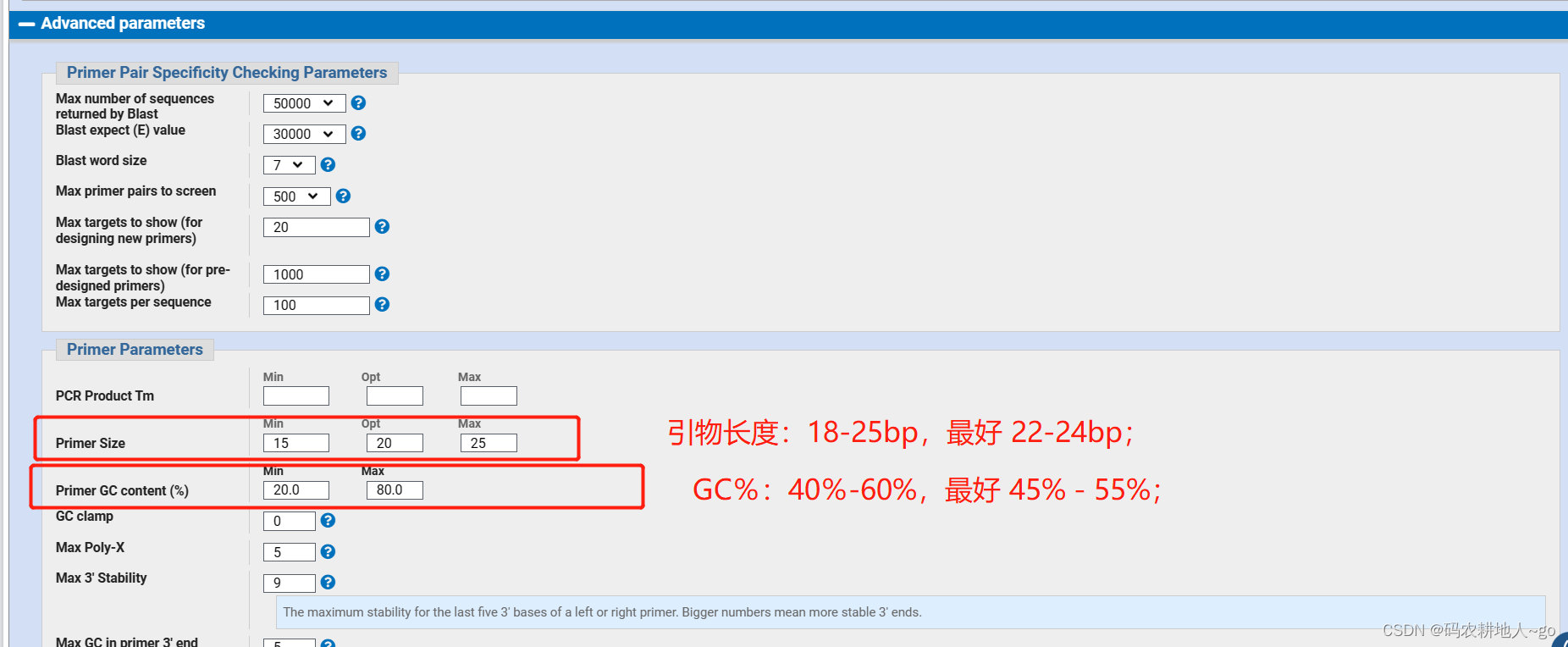
【经验分享】NCBI设计引物——最详细说明及教程!!!适合0基础~
引物设计参数 红色的表示需要改 绿色的表示不需要改 箭头表示普通PCR 接下来,我们需要简单地为引物设计设定一些参数 1. 输入目的模板: 2. 引物位置和产物大小(qPCR可以设置跨越两端的外显子的长度,PCR也可以控制左右引物位置) 需要要求产物长度80--200bp,太短或者太长都不适合做荧光定量PCR检测,特别是荧光定量PCR的引物,要控制TM=60℃左右。 3.
linux下载sra数据库,从NCBI当中SRA数据库中下载高通量测序数据
用别人的数据,发自己的文章 由于大多数杂志在文章发表前要求公开数据,所以随着测序文章的爆发,NCBI的SRA数据库当中积累了海量的测序数据。我们可以利用这些数据重新做数据挖掘,发表新的文章。 官方下载方法不太稳 要利用数据,首先得下载得到数据,虽然SRA数据库提供的SRA Toolkit 工具包里的prefetch可以下载,但是用这个方法下载数据需要经过复杂的设置,而且经常莫名奇妙的下不了,总的
linux下载sra数据库,从NCBI当中SRA数据库中下载高通量测序数据
用别人的数据,发自己的文章 由于大多数杂志在文章发表前要求公开数据,所以随着测序文章的爆发,NCBI的SRA数据库当中积累了海量的测序数据。我们可以利用这些数据重新做数据挖掘,发表新的文章。 官方下载方法不太稳 要利用数据,首先得下载得到数据,虽然SRA数据库提供的SRA Toolkit 工具包里的prefetch可以下载,但是用这个方法下载数据需要经过复杂的设置,而且经常莫名奇妙的下不了,总的
Linux系统中已知SRR号如何从NCBI上下载SRA数据到服务器中
高通量组学数据目前已经成为生物研究的重要板块,对于一些文章中出现的数据的挖掘尤其是人体数据的再利用也成为探究科学问题的重要前沿组成。通常情况下文章的高通量数据需要上传到NCBI的SRA(Sequence Read Archive)供大家下载学习,而我们也可以通过多种方法对数据进行下载再挖掘。在此介绍一种下载NCBI SRA数据的最佳方法。 首先,我们在下面的网址中
ncbi-genome-download批量下载基因组数据
1. ncbi-genome-download 的下载和安装 ncbi-genome-download 是一个可以直接从NCBI上批量下载序列的软件,支持下载多种格式 利用 conda 对其直接安装 参考 #创建环境conda create -n ncbi_genome_download#激活环境conda activate ncbi_genome_download# 安装cond