fasta专题

fasta文件读取c++代码
#include <iostream>#include <fstream>#include <string>#include <vector>using namespace std;// 读取FASTA格式数据并存储到字符串向量中void readFasta(const string& filename, vector<string>& sequences, vector<strin
![[python项目一]查找输出fasta序列的gap的起始终止等信息](https://img-blog.csdn.net/20151008112525570?watermark/2/text/aHR0cDovL2Jsb2cuY3Nkbi5uZXQv/font/5a6L5L2T/fontsize/400/fill/I0JBQkFCMA==/dissolve/70/gravity/Center)
[python项目一]查找输出fasta序列的gap的起始终止等信息
一、需要实现的程序内容及输出: 对于输入的fasta序列,编写程序查找里面N的起始,终止位置等信息,如下面的染色体test.fa序列为例: >1 dna_sm:chromosome chromosome:UMD3.1:1:1:158337067:1 REF aaattagacactgaagagacttggaaagagaggaagtcaaataacaaagaagaggaaacc aaaagggc
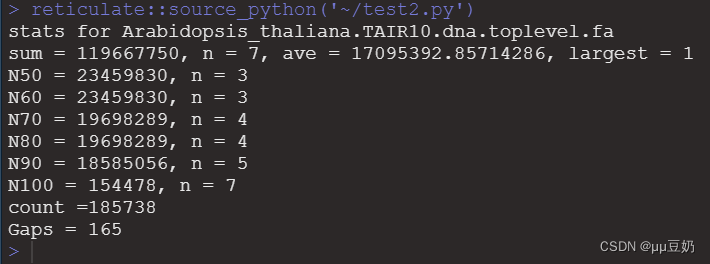
从基因组获取fasta文件并计算Nx0脚本
从基因组获取fasta文件并计算Nx0脚本 import sysfrom Bio import SeqIO from Bio.SeqRecord import SeqRecordfrom Bio.Seq import Seqimport re #正则表达式 我们先导入模块,sys将参数提取到脚本外,实现把路径跟在脚本后面直接跑即可。SeqIO是Bio里面的子包,用于序列信息提取,re
FASTA序列格式说明
fasta序列格式是blast组织数据的基本格式,无论是数据库还是查询序列,大多数情况都使用fasta序列格式,所以首先对fasta格式在做详细说明。 下面是一个来源于NCBI的fasta格式序列: >gi|187608668|ref|NM_001043364.2| Bombyx mori moricin (Mor), mRNAAAACCGCGCAGTTATTTAAAATATGAATATTT

从NBCI上下载大量细菌的SRA测序文件及其对应的fasta文件
第一步 从genome数据库中下载对应信息 打开NCBI,进入genome数据库从Custom resources下进入Microbes,再进入Browse microbial genomes输入需要的物种,可以进一步筛选(filter),然后download 第二步 提取csv结果文件中的biosmple列的biosample号和replicon列的chromesome号 replicon
蛋白质FASTA与药物分子SMILES数据集文本数据处理与可视化分析(一)2021SC@SDUSC
本文基于davis数据集进行操作2021SC@SDUSC 首先最直接的方式是学习一下别人的项目中是如何处理数据的 在项目代码中,指定了一个ParamList的字典用键值对的方式存储配置信息 下图为所选的数据集ESOL_SMILESValue.txt的结构,每一行数据逗号左侧为分子SMILES序列,右侧为该分子对应的label 下一步就是把刚刚的ParamList(此次已封装到opt里),

根据id提取fasta序列
Perl脚本练习 bioperl读入写出fasta 要求 根据序列ID,从fasta文件中提取目标序列并输出 数据 序列ID fasta文件 思路 以序列ID为键,构建哈希用bioperl读入fasta,获得序列id如果id存在于哈希中,输出序列 代码 die "perl $0 <id> <fa> <OUT>" unless(@ARGV==3);#$0程序名use Bio::
pymol安装使用;vscode蛋白质可视化插件 protein viewer;rcsb pdb,fasta蛋白wget下载
pdb文件字段说明学习: 1、pymol安装使用 参考:https://blog.csdn.net/eternalapple/article/details/110263296 官网:https://pymol.org/ 安装:1、可以直接conda安装,一条命令conda install -c schrodinger pymol-bundle安装成功后输入pymol即可调出窗口
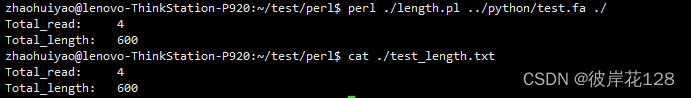
perl语言——length.pl脚本(统计fasta文件序列长度)
Perl脚本——stat.pl(统计fasta文件序列长度) 相比Perl语言,现在python用的多。但是perl依旧是生信学习的一门课程,还是有人在写,所以你至少要会读。 #!/use/bin/perl #perl解析器$inputFile = $ARGV[0]; #输入文件:fasta$outputDir = $ARGV[1]; #输出目录if (@ARGV