本文主要是介绍[python项目一]查找输出fasta序列的gap的起始终止等信息,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
一、需要实现的程序内容及输出:
对于输入的fasta序列,编写程序查找里面N的起始,终止位置等信息,如下面的染色体test.fa序列为例:
>1 dna_sm:chromosome chromosome:UMD3.1:1:1:158337067:1 REF
aaattagacactgaagagacttggaaagagaggaagtcaaataacaaagaagaggaaacc
aaaagggcctatagaccttgagtattctcaaggtggaacaagaaactatctgaaattgaa
ccgacccccacgctgcccacaacagctccagagaaattcctagatatatttttactacta
tcataaAAAAAatgattgagtttattttgtatttttaatattgtatttttgagagtgtat
cttctctacttcactctgtgaatctctaggtgttctgggctgtggagaacacttagggaa
>2
ctgattactggctagatcagtctctccccttttgtttgcccttcttctcctcctggtcac
tccaaaacttgagaacaccaggaaactcctgactccaggaacattaatcaacaagagctc
atccaaaagcctccatacctacacggaaaccaagctccatccaagagccaacaagttcca
NNNTCTTTTGACTCTCCCTTTTCTCTCCCATGTCAGCTCTTTCTCCTCCCTCCCCCTTCT
gatcaagacataccatgctaattctccaacaacataggaacatagccctgaacattaaaa
tacaggctgcccaacgtcatgtcaaacccatagatgccccaaaactcactcctggacact
>3
tcattgcactccagagagaagagatccagttccaccgaccagaacacagatgcaagtttc
caaacccaatcaaaagaggaagagatagggagtctacctgaaaaagaattcagagtaatg
>4
gatcaataatgaataatgcaataacagatcaaaagaactctggagggaaacaacagtaga
ggcatgagaaaatacctgaggagataatagttgaaattttctctaaaatggggaaggaaa
atcctaagacatacattaatcaaattaatgaagaccaaacacaaagaacaaatattaaag
TTTTTTTTAATAAATGCCAATCTGTTTATGACTTAACTTGTCANNNNNNNNNNNNNNNNN
NNNNNNNNANNCCCTNNNNNNNNACTTCAGACAATAATGTTTTTTTAAAACCAGTCTAGT
TTCTTGGACTTCTAGTTGGATGGCTTCACCGACTTGAAGGACGTGAGTTTGAGTAAGTTC
CAAGAGTTAGTGATGGACAGGGAAGCCCGGTGTGCTGCAGTCCCTGGGGTGGCAAAGAGT
希望得到每一条染色体N的pos的起始位置,终止位置,长度以及中的Gap(N又称为Gap区域)的总长及总数目,输出结果为:
test.fa.pos:
ID=>2 180 182 3
ID=>4 223 247 25
ID=>4 249 250 2
ID=>4 255 262 8
test.fa.stat:
Total_gap_num=4,Total_gap_len=38
二、用perl的相应的程序如下:
#/user/bin/perl -w
use strict;
unless(@ARGV==1){
die"Usage:perl $0 <input.fa>\n";
}
my($infile)=@ARGV;
open IN,$infile||die"error:can't open infile:$infile";
my $outfile1=$infile."_out";
my $outfile2=$infile."_stat";
open OUT,">$outfile1"||die$!;
open OUTT,">$outfile2"||die$!;
$/=">";<IN>;
my $start=0;
my $skip=0;
my $step;
my $len=1;
my $stop;
my $end;
my $total_len=0;
my $number=0;
my $num_1bp=0;
my $line;
my $i;
while(my $seq=<IN>)
{
if(index($seq,"N")!=-1)
{#if-1
my $id=$1 if($seq=~/^(\S+)/);
chomp $seq;
$seq=~s/^.+?\n//;
$seq=~s/\s//g;
if(index($seq,"N")==-1)
{
last;
}
$step=0;
$stop=1;
$start=index($seq,"N",$step)+1;
$step=$start-1;
$skip=$step;
print "start=$start\tstep=$step\tskip=$skip\n";
while($stop)
{#while -2
$skip=index($seq,"N",$step+1);
print "in while:skip=$skip\tstep=$step\n";
if($skip==($step+1))
{#if skip (49)
print "in-while-if:skip=$skip\tstep=$step\n";
$len++;
$step++;
next;
}else{
print "in-while-else:skip=$skip\tstep=$step\n";
if($skip!=-1)
{#if skip != -1 (55)
print "else-if:skip=$skip\tstep=$step\n";
if($len!=1){
$end=$start+$len-1;
}
else{
$num_1bp++;
$end=$start;
}
$total_len+=$len;
$number++;
print OUT"if-$id\t$start\t$end\t$len\n";
$step=$skip;
$start=$skip+1;
$len=1;
}else{
print "else-else:skip=$skip\tstep=$step\n";
if($len!=1){
$end=$start+$len-1;
}
else{
$num_1bp++;
$end=$start;
}
$total_len+=$len;
$number++;
print OUT"else-$id\t$start\t$end\t$len\n";
$stop=0;
$len=1;
}#if-else- (56)
}#if-else- (49)
}#while -2
}#if-1
}#while
print OUTT "total_length\t $total_len\ngap_number\t$number\n1bp_gap_number\t$num_1bp\n";
$/="\n";
close IN;
close OUT;
三、用python编写的程序如下:
#-*- coding=utf-8 -*-
#输出gap的起始位置,终止位置,长度等位置信息
import os,sys
import re
class Fasta():
def __init__(self,name,sequence):
self.name=name
self.sequence=sequence
def process_fasta(infile):
reader=infile.readlines()
index=0
increace=[]
for line in reader:
line=line.strip()
if line.startswith('>'):
if index >=1:
increace.append(instance)
id=line
seq=''
index += 1
else:
seq += line
instance=Fasta(id,seq)
increace.append(instance)
return increace
def find_N(List):
gap_num=0
gap_len=0
for t in List:
str1=t.sequence
start=0
end=0
length=1
if(str1.find('N') != -1):
indel = str1.find('N',start)
start = indel
step = start
skip = step
flag=True
N_array=re.split('[N+]{1,}',str1)
while(flag):
skip=str1.find('N',step+1)
if(skip - 1 == step):
#前后两次查找的N的index相差1,说明N是连续的
step += 1
length +=1
continue
else:
#前后两次index相差不为1,说明N之间出现了其他碱基
if (skip != -1):
#说明后面还有N
end = bool(length !=1) and start+length -1 or start
step=skip
outfile1.write("ID=%s\t%d\t%d\t%d\n" %(t.name,start,end,length))
length=1
start=step
else:
#说明后面已经没有N了
end = bool(length !=1) and start+length -1 or start
outfile1.write("ID=%s\t%d\t%d\t%d\n" %(t.name,start,end,length))
length=1
flag=False
gap_len += str1.count('N')
gap_num += len(N_array) -1
outfile2.write("Total_gap_num=%d,Total_gap_len=%d\n" %(gap_num,gap_len))
if __name__ == '__main__':
infile=open(sys.argv[1],'r')
outfile1=open(sys.argv[1]+'.pos','w')
outfile2=open(sys.argv[1]+'.stat','w')
List=process_fasta(infile)
find_N(List)
infile.close()
outfile1.close()
outfile2.close()
四、总结:
编程思路请见下面的逻辑图
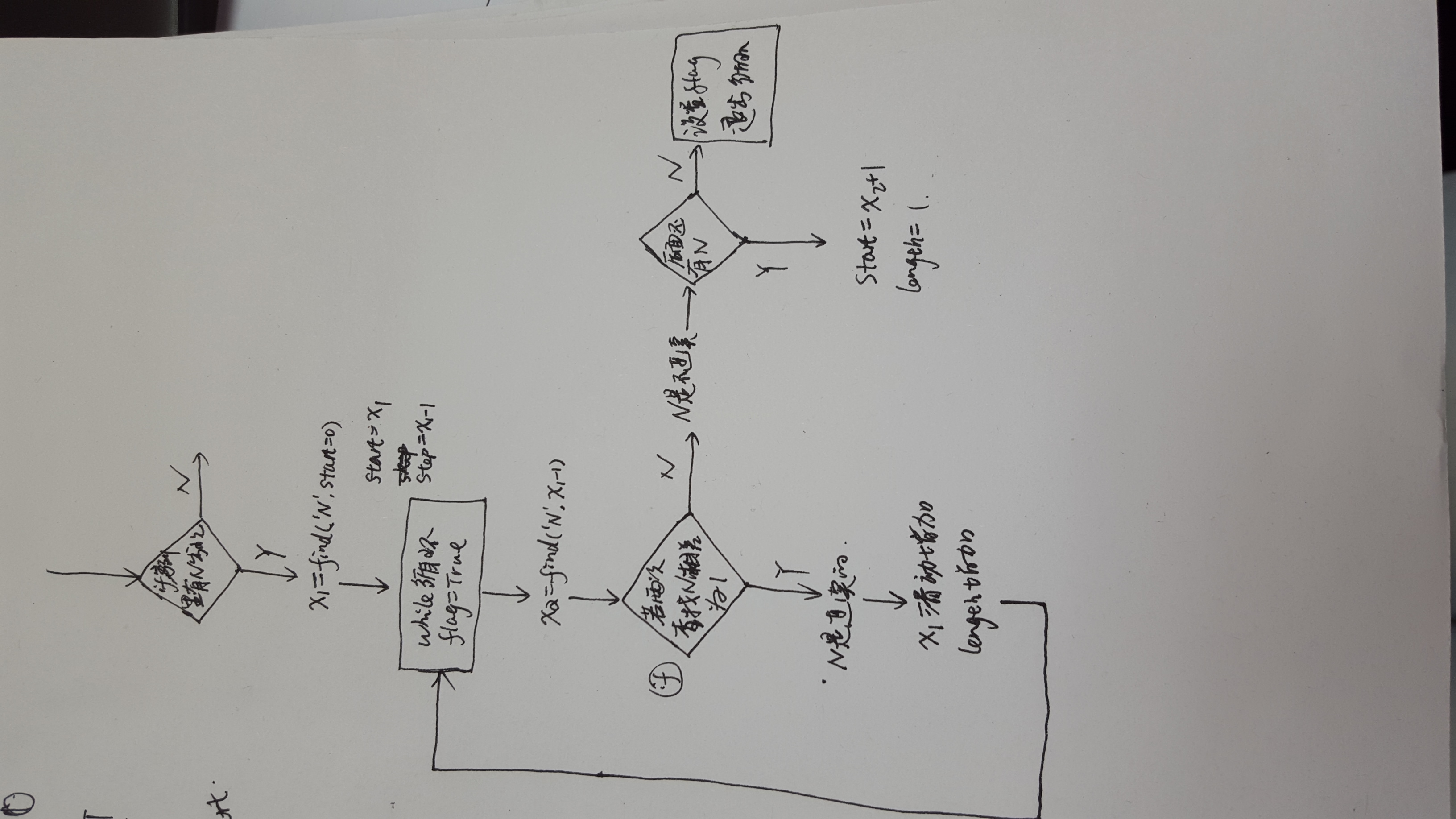
这篇关于[python项目一]查找输出fasta序列的gap的起始终止等信息的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!





