本文主要是介绍Gromacs模拟一:配体-双链蛋白质复合物体系准备,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
1、蛋白质的准备:
在RCSB网站下载想要的蛋白晶体(教程里是3htb),用notepad等编辑器或是分子可视化软件除去里面的非蛋白分子或离子。
这里采用的是一个经过分子对接后的蛋白质pdb和配体小分子的pdb。教程里提到的配体是2-丙基苯酚(JZ4),是一个非肽类分子,同时存在于3htb蛋白里。可以用notepad给他抽出为一个单独的pdb。如果是一个短肽类分子,可以利用gromacs自带的蛋白力场为其生成力场参数(应该可以)。
将蛋白质pdb进行处理,生成top文件和gro坐标文件,命令运行后弹出的力场选项中选择第1个CHARMM36力场:
gmx pdb2gmx -f 3HTB_clean.pdb -o 3HTB_processed.gro -ter之后,选择TIP3P模型作为水分子,选择NH3+和COO-作为蛋白链的末端原子。
2、将配体分子转为需要的mol2格式:
我还是用discovery studio进行加H(添加力场)和输出为mol2文件。
下载相关的charmm36力场charmm36-jul2022.ff.tgz和一个python脚本cenff_charmm2gmx.py,根据实际环境。下载链接http://mackerell.umaryland.edu/charmm_ff.shtml#gromacs
将下载的tgz文件解压到存放蛋白和配体pdb文件的工作目录下,成为一个子目录。
3、为配体mol2文件生成拓扑文件:
在CGenFF 服务器注册账号,然后使用这个网站完成拓扑数据的生成,但是我注册不了(填好注册信息,点击注册,卡,信息清空..)
下载Sob老师提供的Sobtop程序可以gromacs适合的拓扑文件,同时这个程序非常方便,下载后打开里面的exe文件就能运行(大佬不愧是大佬)。
下载界面的下方有11个例子参考,我根据例子2完成对小分子拓扑文件的生成。

4、建立复合物的gro文件:
用protein.gro拷贝成一个新文件,修改名为complex.gro。将ligand.gro里的原子坐标放到complex里,再在盒子数据上方空一行:
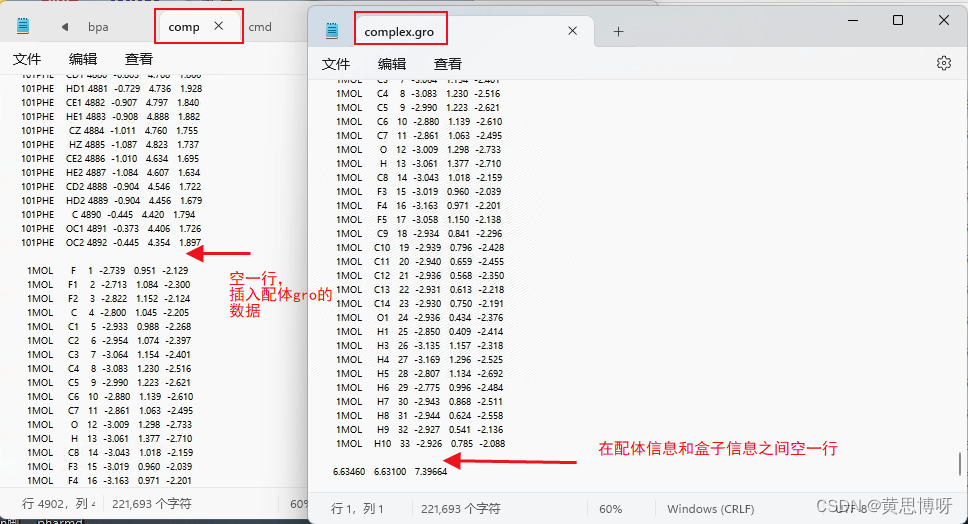
这里推荐使用notepad3进行编辑,win10自带的笔记本确实不行,不能看清楚数据之间的空格情况,容易报错。
这里不用留空行,留了会报错。
主要的问题应该还是空格没有处理好。蛋白有4892个原子,配体是33个原子,所以在complex.gro的第2行(表示原子数)修改为4825
配体里的原子数从1又重新排序了,这个应该不伤大雅。
5、建立复合物的拓扑文件:
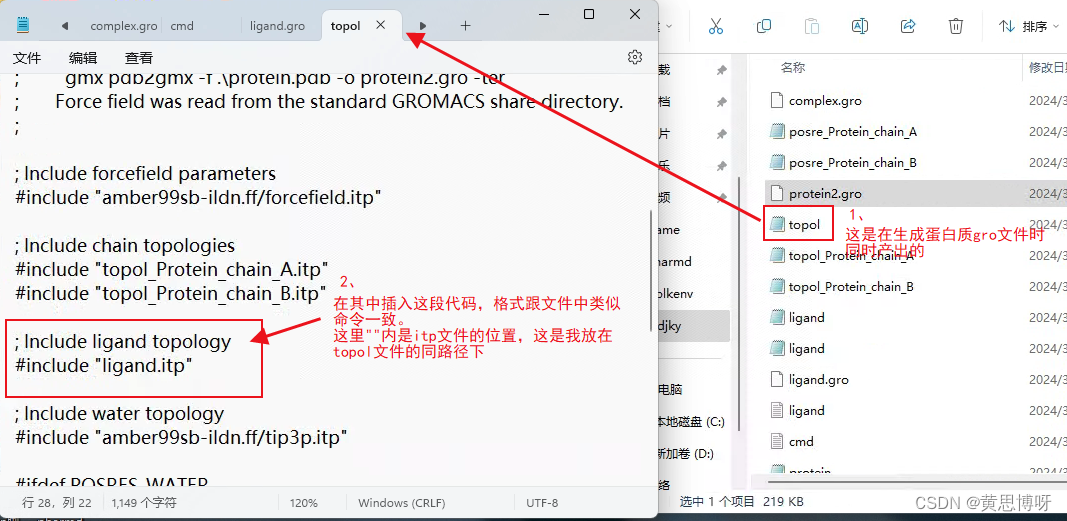
同目录下有个topol,是一个包含蛋白质拓扑信息的文件(在使用pdb2gmx时生成)。这个蛋白是一个双链蛋白,因为活性口袋在两个蛋白双螺旋之间。同目录下还有两个分开的独立链的top文件。
在topol里,用#include语句导入蛋白的top信息,如果蛋白是单链的话,数据会直接导入到topol中。根据前面导入力场,导入蛋白链拓扑的命令的格式,插入导入配体拓扑的命令:
6、规避报错,关于[atomtypes]:
利用Sob老师生成的小分子itp文件中,有[atomtypes]这个部分,如果不处理,会报错。确实在官方manual里的top文件模板中,不会出现[atomtypes]这一项。
在manual里,top文件的格式如下,是没有[atomtypes]这个部分的。
;
; Example topology file
;
[ defaults ]
; nbfunc comb-rule gen-pairs fudgeLJ fudgeQQ1 1 no 1.0 1.0; The force field files to be included
#include "rt41c5.itp"[ moleculetype ]
; name nrexcl
Urea 3[ atoms ]
; nr type resnr residu atom cgnr charge1 C 1 UREA C1 1 0.6832 O 1 UREA O2 1 -0.6833 NT 1 UREA N3 2 -0.622[ bonds ]
; ai aj funct c0 c13 4 1 1.000000e-01 3.744680e+053 5 1 1.000000e-01 3.744680e+056 7 1 1.000000e-01 3.744680e+05[ pairs ]
; ai aj funct c0 c12 4 1 0.000000e+00 0.000000e+002 5 1 0.000000e+00 0.000000e+002 7 1 0.000000e+00 0.000000e+00[ angles ]
; ai aj ak funct c0 c11 3 4 1 1.200000e+02 2.928800e+021 3 5 1 1.200000e+02 2.928800e+024 3 5 1 1.200000e+02 3.347200e+02[ dihedrals ]
; ai aj ak al funct c0 c1 c22 1 3 4 1 1.800000e+02 3.347200e+01 2.000000e+006 1 3 4 1 1.800000e+02 3.347200e+01 2.000000e+002 1 3 5 1 1.800000e+02 3.347200e+01 2.000000e+00[ dihedrals ]
; ai aj ak al funct c0 c13 4 5 1 2 0.000000e+00 1.673600e+02; Include SPC water topology
#include "spc.itp"[ system ]
Urea in Water[ molecules ]
Urea 1
SOL 1000我的做法是将这些原子添加到力场文件中。
如图,根据topol里提供的信息,找到gromacs提供的所用力场的文件地址:
(PS:这里图截错了,实际应该是#include "amber99sb-ildn.ff/forcefield.itp")
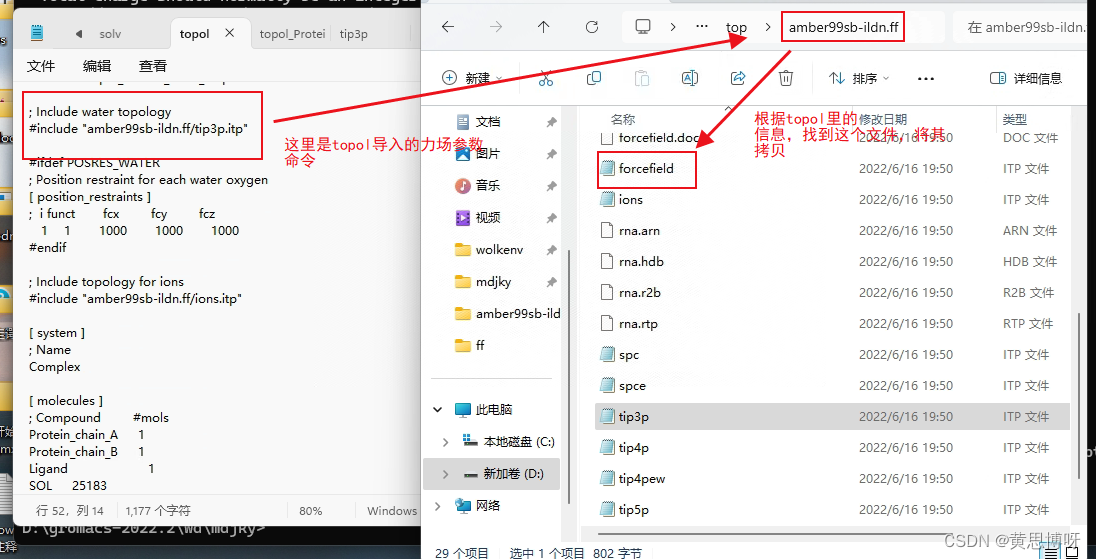
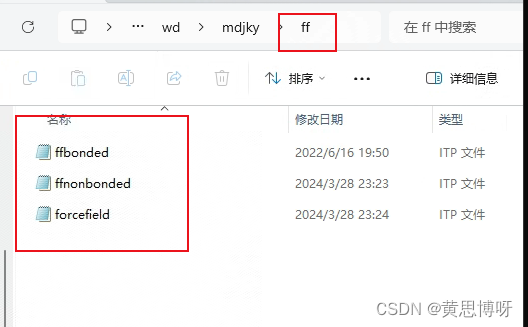
打开forcefield.itp文件,会发现其还是用include命令调用了两个itp文件。 将这三个itp文件拷贝到我的工作目录(存放了protein.gro、ligand.gro、topol什么的文件夹)下的子文件夹ff里:
在ffnonbonded.itp文件里有原子类型信息(用笔记本打开查看),可以将配体itp里的atomtype的数据拷贝过去,最后保存。
之后,修改topol里调用forcefield.itp的命令,将其路径改为调用修改后的forcefield.itp文件存放的位置ff文件夹:

到这里,提示拓扑文件里有[atomtypes]的fatal报错就会消失。
7、定义盒子大小和添加溶剂分子:
定义盒子:
gmx editconf -f complex.gro -o newbox.gro -bt dodecahedron -d 1.0
#要查看每个参数是什么意识,可以用gmx editconf -h添加溶剂水:
gmx solvate -cp newbox.gro -cs spc216.gro -p topol.top -o solv.gro
#没有tip3p.gro
#用spc216的gro代替,不过拓扑信息还是用tip3p的水我觉得不一定要用tip3p.gro,不会报错就不错了![]() 。
。
8、规避报错,分子数量的报错:
这里添加了水到复合物的gro文件里,但是topol文件里的[melocules]还是没有更新的状态,所以有下面的报错:
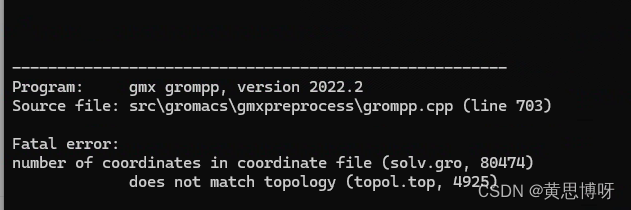
这里提示了坐标文件中原子数和拓扑文件中的原子数不同。Fatal error: number of coordinates in coordinate file (solv.gro, 29510) does not match topology...-CSDN博客
这篇博客中老师的方法给了我提醒。要在topol文件的molecules里添加水的分子数,这里多出75549个原子,就是25183个水分子(除以3)。
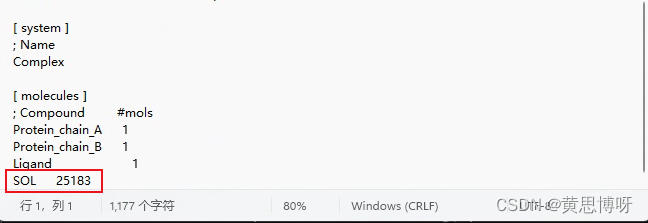
这里为什么是SOL, 其实,这个记录在每个分子的itp文件的[moleculetypes]部分里。如下图,在[dir]\gmx\share\gromacs\top\amber99sb-ildn.ff文件夹下的tip3p.itp文件里的[moleculetype]:
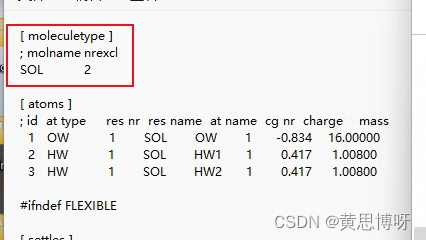
所以,这个水的分子名是SOL。
9、添加离子:
写一个mdp文件,重命名为ions.mdp,真不懂为什么加离子就要用到这种文件。
#生成一个tpr文件:
gmx grompp -f ions.mdp -c solv.gro -p topol.top -o ions.tpr
#添加离子,生成一个新的gro文件:
gmx genion -s ions.tpr -o solv_ions.gro -p topol.top -pname NA -nname CL -neutral# 运行最后一个添加离子的命令后,会出现如下的选项:
Reading file ions.tpr, VERSION 2022.2 (single precision)
......
Group 8 ( SideChain) has 3352 elements
Group 9 ( SideChain-H) has 1174 elements
Group 10 ( Prot-Masses) has 4892 elements
Group 11 ( non-Protein) has 75582 elements
Group 12 ( Other) has 33 elements
Group 13 ( MOL) has 33 elements
Group 14 ( Water) has 75549 elements
Group 15 ( SOL) has 75549 elements
Group 16 ( non-Water) has 4925 elements
Select a group:
#这里输入15输入15,因为我的topol里的溶剂分子类型写的就是SOL,查看topol文件,添加的离子文件已经更新到文件里:

采用VMD或是Pymol检查配体和蛋白之间是否存在挨太近等不合理的状况:
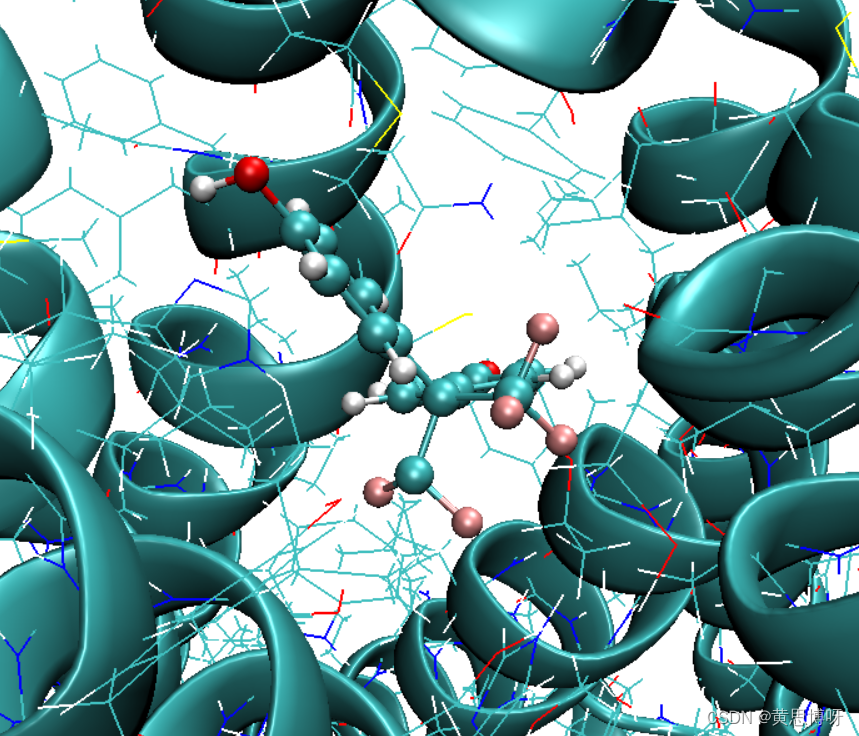
参考教程:
Protein-Ligand Complex
这篇关于Gromacs模拟一:配体-双链蛋白质复合物体系准备的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!









