gromacs专题
Gromacs软件进行蛋白-配体体系MD模拟
1、进行能量最小化 下载官方提供的参数文件:em.mdp ,或者直接复制这里: ; LINES STARTING WITH ';' ARE COMMENTStitle = Minimization ; Title of run; Parameters describing what to do, when to stop and what to saveintegrator

Gromacs——教程学习(6)
谈谈怎么判断分子动力学模拟是否达到了平衡 在计算RMSD之前必须先通过最小二乘法将各帧结构相对于参考结构进行最大程度叠合,从而消除体系的整体运动而令RMSD只体现生物分子内部结构的变化,这称为align或者least squares fit。 需要注意的是,蛋白质骨架的RMSD曲线并不总能反映出蛋白结构特征变化的全貌。一方面,RMSD的计算涉及到一大堆原子的平均,若一个很大的蛋白某个局部出现一

Gromacs——教程学习(4)
分子动力学(MD)模拟,模拟体系构建经验总结 在一个完整的分子动力学模拟中,一般包括以下几个步骤: 1.选择将要使用的力场,并根据模拟体系确定力场参数,构建力场文件; 2.产生初始构型,搭建模拟体系; 3.模拟退火(simulated annealing); 4.平衡体系; 5.模拟数据采样; 6.数据分析、处理。 产生初始构型经验总结 1.产生初试构型的一般步骤是: 1) 给出分子内
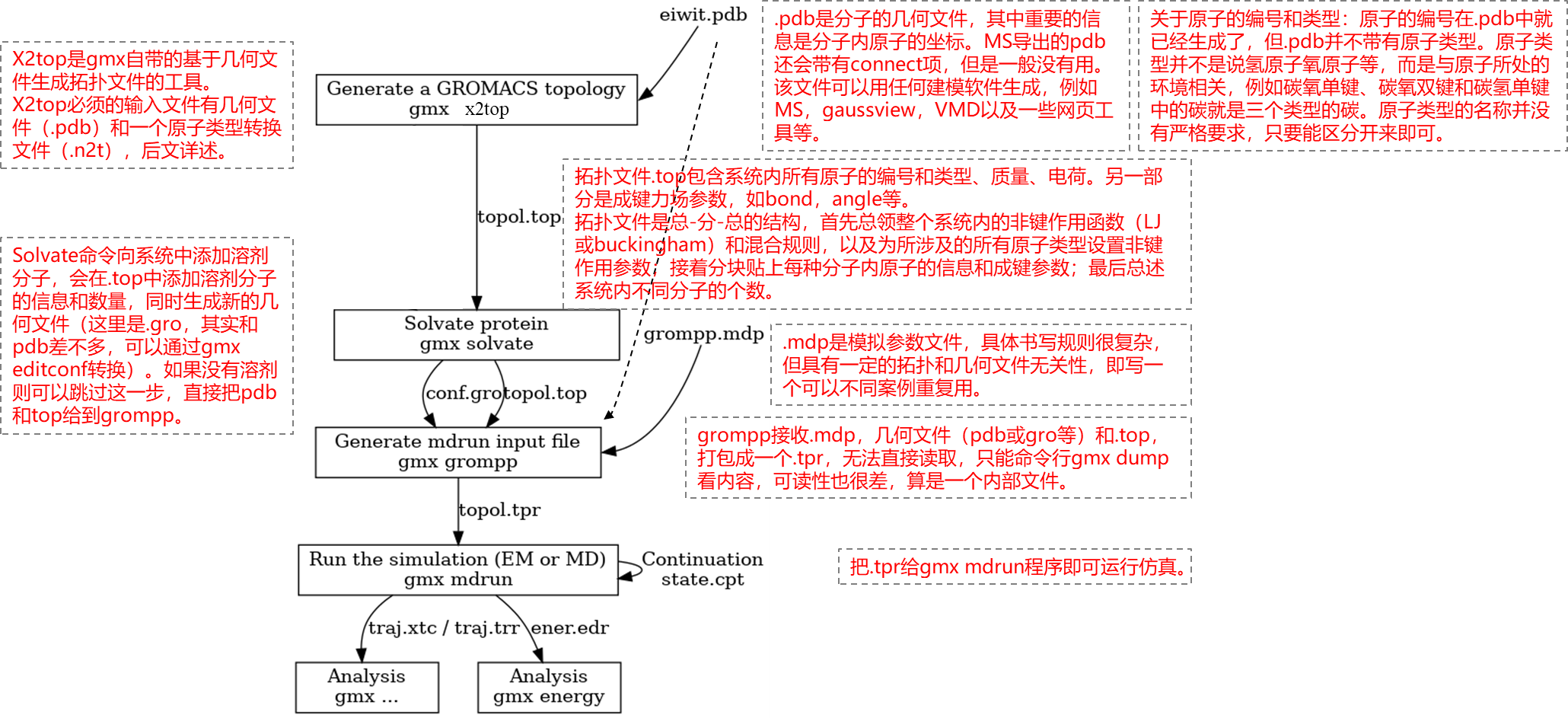
分子动力学模拟学习-Gromacs工具链
1、总体流程 在gromacs的使用说明中有一个flow chart,比较简略。以下针对一般体系(非蛋白等领域)进行了一些调整,通用性更强。 在做分子动力学模拟时,其复杂性除了以上各种输入输出文件的操作,另一点就是力场参数、原子电荷数据的获取。从本质上讲,MD模拟软件就是读取系统内原子的位置、质量、电荷、成键和非键关系,在不同的环境条件下进行计算,而我们做的就是正确地构建输入文件。 2、
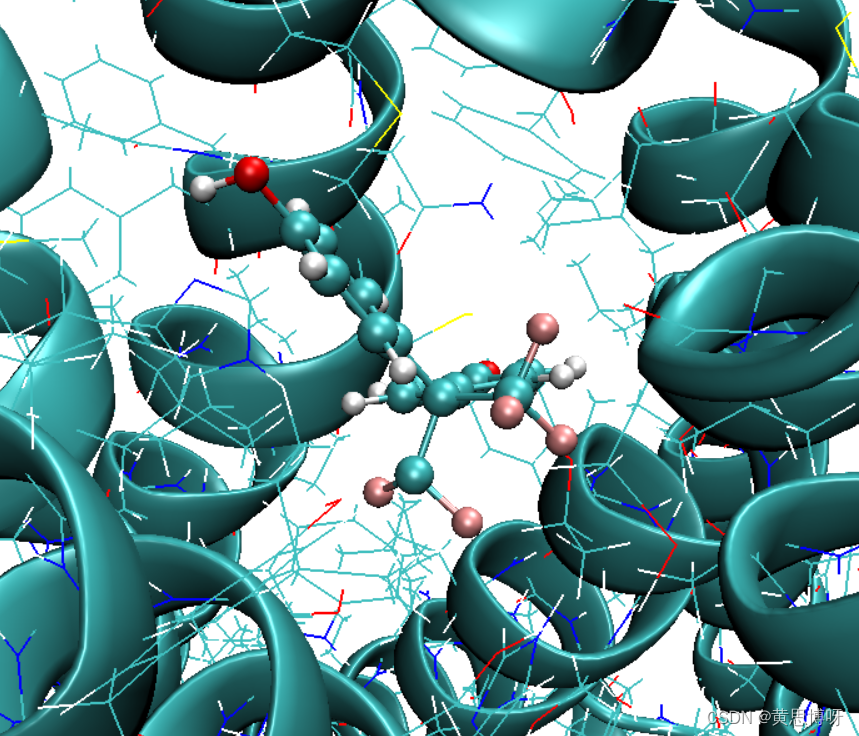
Gromacs模拟一:配体-双链蛋白质复合物体系准备
1、蛋白质的准备: 在RCSB网站下载想要的蛋白晶体(教程里是3htb),用notepad等编辑器或是分子可视化软件除去里面的非蛋白分子或离子。 这里采用的是一个经过分子对接后的蛋白质pdb和配体小分子的pdb。 教程里提到的配体是2-丙基苯酚(JZ4),是一个非肽类分子,同时存在于3htb蛋白里。可以用notepad给他抽出为一个单独的pdb。如果是一个短肽类分子,可以利用gromacs自
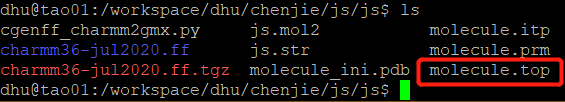
GCenFF生成Gromacs Topol文件用于CHARMM36力场
这是自己初学Gromacs后,第一次自己用GCenFF生成Gromacs topol 文件。此文作为自己的学堂札记便于日后回访。同时分享给刚入门Gromacs分子动力学模拟的同学。 环境准备: Linux装有1. numpy; 2. networkx (1.11); Python 2.7.3 或Python 3.5.6 GCenFF生成topol文件整体流程如下: 1. 拿Gaussian
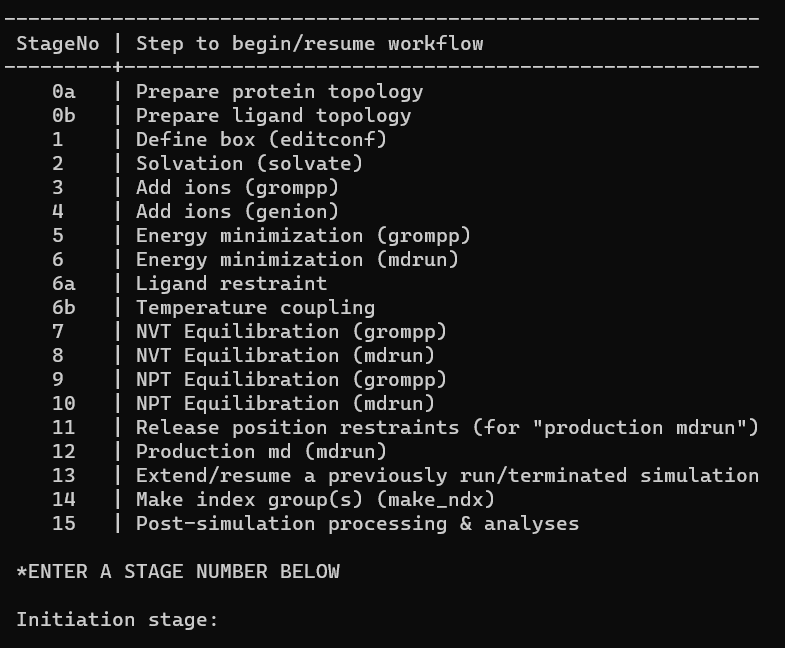
CHAPERONg:基于GROMACS的分子动力学模拟和轨迹分析的自动化工具
此工作介绍了一个名为CHAPERONg的工具,它是一个自动化的GROMACS(一种广泛使用的分子动力学模拟软件)模拟和轨迹分析工具。CHAPERONg旨在简化和自动化蛋白质和蛋白质-配体系统的GROMACS分子动力学(MD)模拟流程,包括常规MD模拟、拉伸MD(steered MD)和增强型伞采样(umbrella sampling)模拟。这个工具通过集成GROMACS模块和第三方工具,提供

gromacs + plumed 编译安装教程
简介 gromacs的基本编译安装教程请参考:gromacs 编译安装教程。 本文介绍如何结合plumed安装gromacs。 编译plumed PLUMED 是分子系统中自由能计算的开源库,与一些最流行的分子动力学引擎一起工作。 可以使用诸如元动力学,伞形采样和基于 Jarzynski 方程的转向 MD 之类的现有技术来执行自由能计算作为许多次序参数的函数,特别关注生物学问题。 官网地
Gromacs 布朗动力学模拟 Gromacs安装教程 虚拟机安装 VMware Workstation Pro任选版本 Linux CentOS7 Ubunto Xshell和Xftp安装
主题简述 Gromacs是研究生物大分子体系的动力学模拟软件,在国内外有着广泛的应用。目前Gromacs只发布了基于Linux/Unix的安装程序包,本教程将介绍在VMware虚拟机上安装Gromacs正式版本2019.6。 工具/原料 VMware 虚拟机容器和Linux系统CentOS7对应镜像文件——官网下载 Cmake和Gromacs程序包——官网下载(Gromacs官网和cma
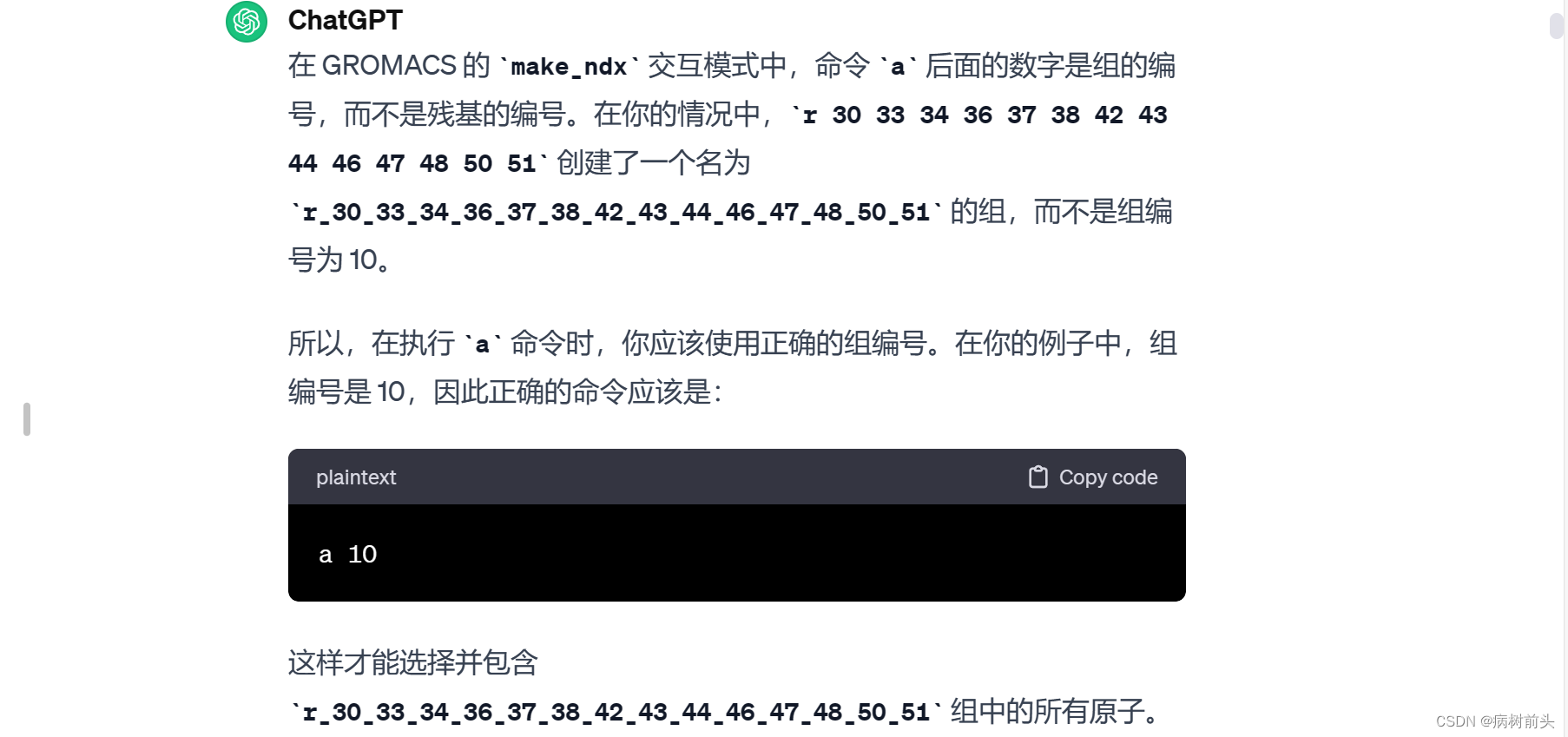
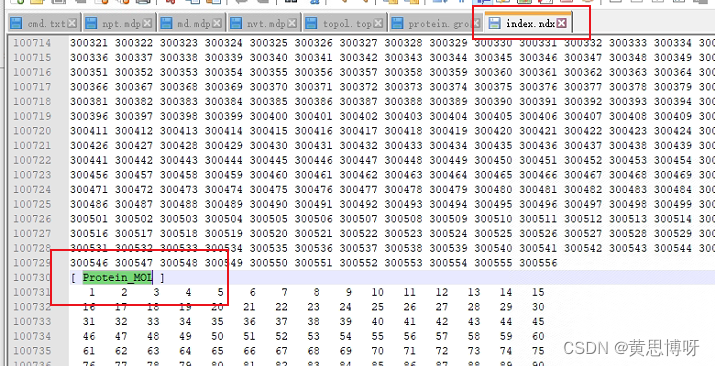
Gromacs make_ndx建组问题
1. 选择特定分子或原子: gmx make_ndx -f input.gro -o output.ndx 这将打开交互式界面,您可以在其中选择要包含在索引文件中的分子和原子。按照提示进行操作,选择适当的分组。 2. 手动创建索引文件: 您还可以手动创建一个文本文件,其中包含要包括在索引文件中的分子和原子的详细信息。然后,您可以使用以下命令: gmx make_ndx -f input.

gromacs学习及使用(2)
命令解释参考GROMACS基本教程 整个流程参考分子动力学模拟Gromacs一般使用步骤(空蛋白) 从gromacs 5.0版本开始,所有的工具都是“gmx”的子模块。可以通过下面的命令获得任何一个模块的帮助信息: gmx help (module) 或者 gmx (module) -h #!/bin/bashmodule load gromacs/gromacs-2021.5-gcc-9
Gromacs基于OPLS-AA力场的聚合物建模及模拟
本文内容包括: OPLS-AA力场简介/高分子模拟常用力场; 聚合物建模方法; Gromacs添加非标准残基建模聚合物的方法; 聚合物PDB文件生成; 开始模拟。 第一步: 尽管Gromacs与Amber都包含适合蛋白质与核酸体系模拟的力场,但到目前为止罕有专为人工聚合物体系开发的力场。不同于蛋白质/核算具有有限种类的残基,聚合物原则上可以有无限种不同的单体,去为每一种单体都设定
SailVina 使用教程 Autodock Vina分子对接全套整合软件 MGLTools闪退 作用力分析 MOE 薛定谔 Gromacs 全网最全分子对接教程
目录 1. 介绍2. 安装SailVina2.1.使用Python运行2.2 直接运行exe程序 3. 对接常用操作教程3.1 获取受体3.1.1 通过SailVina进行获取3.1.2 从pdb网站获取 3.2 准备受体3.2.1 使用SailVina自动准备受体3.2.2 使用ADT手动准备受体 3.3 准备对接位点3.3.1 使用SailVina根据受体中共晶的配体来自动生成对接位点3
Gromacs的使用的
第一章 Gromacs的安装 一、Grommacs Gromacs是分子动力学模拟中必不可少的工具,需要注意的是在MD的模拟中是不涉及化学键的建立和生成的,它停留在原子的键角,键长,范德华力和库仑力的平面上,如果你想探究原子之间成键的现象以及电子云的运动,请把注意力放在Gaussian上来。其次Gromacs是运行在Linux系统上的,他的操作一般以命令的形式进行。所以你首先要拥有

2023-CADD、AIDD/分子对接、gromacs分子动力学全程实操、代谢组学
CADD(Computer Aided Drug Design):计算机辅助药物设计,依据生物化学、酶学、分子生物学以及遗传学等生命科学的研究成果,针对这些基础研究中所揭示的包括酶、受体、离子通道及核酸等潜在的药物设计靶点,并参考其它类源性配体或天然产物的化学结构特征,以计算机化学为基础,通过计算机的模拟、计算和预算药物与受体生物大分子之间的相互作用,考察药物与靶点的结构互补、性质互补等,设计出合