本文主要是介绍Gromacs软件进行蛋白-配体体系MD模拟,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
1、进行能量最小化
下载官方提供的参数文件:em.mdp ,或者直接复制这里:
; LINES STARTING WITH ';' ARE COMMENTS
title = Minimization ; Title of run; Parameters describing what to do, when to stop and what to save
integrator = steep ; Algorithm (steep = steepest descent minimization)
emtol = 1000.0 ; Stop minimization when the maximum force < 10.0 kJ/mol
emstep = 0.01 ; Energy step size
nsteps = 50000 ; Maximum number of (minimization) steps to perform; Parameters describing how to find the neighbors of each atom and how to calculate the interactions
nstlist = 1 ; Frequency to update the neighbor list and long range forces
cutoff-scheme = Verlet
ns_type = grid ; Method to determine neighbor list (simple, grid)
rlist = 1.2 ; Cut-off for making neighbor list (short range forces)
coulombtype = PME ; Treatment of long range electrostatic interactions
rcoulomb = 1.2 ; long range electrostatic cut-off
vdwtype = cutoff
vdw-modifier = force-switch
rvdw-switch = 1.0
rvdw = 1.2 ; long range Van der Waals cut-off
pbc = xyz ; Periodic Boundary Conditions
DispCorr = nomdp文件里分3个区块,标题、模拟的步数怎么跑和原子之间相互作用力设置的有关参数。
例如模拟的步数怎么跑里的设置:能量下降的积分算法用最陡下降最小化。体系中最大能量达到1000停止最小化。每步下降能量大小是0.01以及最大的步数设置为50000。
gmx grompp -f em.mdp -c solv_ions.gro -p topol.top -o em.tpr先编译执行文件tpr,然后调用mdrun执行能量最小化:
gmx mdrun -v -deffnm em
#-v 将详细内容输出到终端
#-deffnm em 指定输出和输入文件名都叫em
查看输出的结果:
Steepest Descents converged to Fmax < 1000 in 143 steps
Potential Energy = -4.9014547e+05
Maximum force = 8.7411469e+02 on atom 27
Norm of force = 5.6676244e+01输出的结果上看,算法在第143步就实现体系中最大能量小于1000,然后结束最小化。这里的力是指原子在原位上受到的键作用力和非键作用力的总和。
这里的Maximum force=800 on atom 27说明体系中有最大能量是27号原子。
2、完成能量最小化后,建立一个需要限制的配体原子的信息的文件。
我的配体的gro文件是ligand.gro,在命令行输入:
gmx make_ndx -f ligand.gro -o index_ligand.ndx #进入交互界面> 0 & ! a H* #选择0的system中全部非H原子> q #退出index_ligand.ndx文件内容入下,就是配体原子的原子数据索引:
[ System ] #整个系统的配体原子序号(索引)1 2 3 4 5 6 7 8 9 10 11 12 13 14 1516 17 18 19 20 21 22 23 24 25 26 27 28 29 3031 32 33
......
[ System_&_!H* ] #选出的要限制的非H配体原子序号(索引)1 2 3 4 5 6 7 8 9 10 11 12 14 15 1617 18 19 20 21 22 23 24建立对选出的非H原子带有限制力信息的itp文件(拓扑文件)
gmx genrestr -f ligand.gro -n index_ligand.ndx -o posre_ligand.itp -fc 1000 1000 1000
#生产出itp拓扑文件,-fc指出限制力,对ndx里的选出的非H原子用在笛卡尔坐标中各个轴用1000的力栓住
#该命令会进入交互界面
#选出要限制的原子就是了在topol文件里加入导入限制力参数posre_l.itp文件的命令:

3、进行温度耦合
温度耦合就是对原先没有温度概念的系统附加上温度。对每种分子类型和它自己的恒温基团耦合是一个坏主意。Gromacs的恒温器不适合对小分子单独进行耦合
因为gmx的温度耦合算法不稳定,少量原子组成的组别单独进行单独进行耦合,动能波动会很大,会导致体系的崩溃。例如教程里提到的,如果在nvt.mdp里设置温度耦合的组别是这样:
tc-grps = Protein JZ4 SOL CLSOL是水,Protein是蛋白,这两的原子数目量多(超过1000),单独进行温度耦合是不会有动能波动大的问题的。但是离子Cl-和配体JZ4,体系里CL只有6个,JZ4是22个(教程中的数据),原子数目完全不是体系中蛋白和水分子在一个量级。不能单独进行温度耦合。
JZ4是配体,和蛋白挨得很近,可以将其视为一个整体进行温度耦合,离子则是苟在水分子群里,可以和所有水分子组成一个整体进行耦合。典型的tc-grps设置是:
tc-grps=Protein Non-Protein #典型的设置现在就是要把配体和蛋白整合为Protein,水和离子整合为Non-Protein。
首先,采用make_ndx模块将蛋白质和配体的index抽出,做为一个区块[Protein_JZ4],这个名字是默认的,输出到文件index.ndx里。这个区块的名字要记住,可以在输出的index.ndx里看到。
gmx make_ndx -f em.gro -o index.ndx
....... #下面是选项0 System : 33506 atoms1 Protein : 2614 atoms
......13 JZ4 : 22 atoms
......21 Water_and_ions : 30870 atoms> 1 | 13 #这里选中蛋白和配体, 这里已经存在water_and_ions,就不需要再抽出index。
> q在我的index.ndx文件里存在water_and_ions:
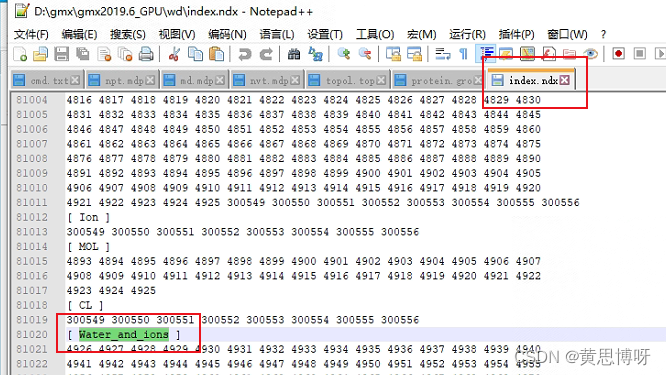
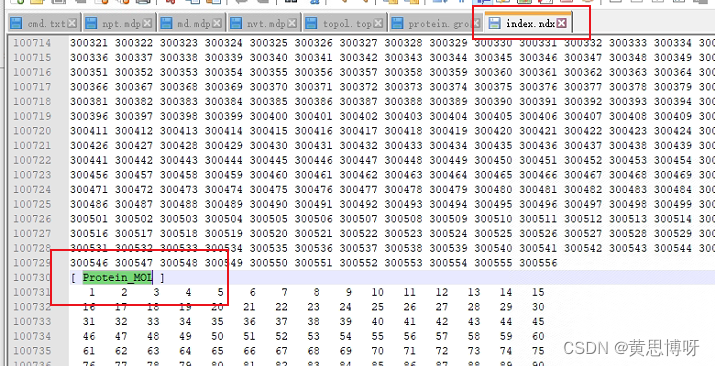
同时也存在蛋白和配体的组和(我的配体是分子名叫MOL,不是JZ4):
下载官方的nvt.mdp文件,或者使用下面的:
注意mdp里的这个设置,要根据自己体系的分子名进行调整:
tc-grps=Protein_JZ4 Water_and_ions #这是这个体系的设置,修改在nvt.mdp里nvt.mdp文件:
title = Protein-ligand complex NVT equilibration
define = -DPOSRES ; position restrain the protein and ligand
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 50000 ; 2 * 50000 = 100 ps
dt = 0.002 ; 2 fs
; Output control
nstenergy = 500 ; save energies every 1.0 ps
nstlog = 500 ; update log file every 1.0 ps
nstxout-compressed = 500 ; save coordinates every 1.0 ps
; Bond parameters
continuation = no ; first dynamics run
constraint_algorithm = lincs ; holonomic constraints
constraints = h-bonds ; bonds to H are constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Neighbor searching and vdW
cutoff-scheme = Verlet
ns_type = grid ; search neighboring grid cells
nstlist = 20 ; largely irrelevant with Verlet
rlist = 1.2
vdwtype = cutoff
vdw-modifier = force-switch
rvdw-switch = 1.0
rvdw = 1.2 ; short-range van der Waals cutoff (in nm)
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
rcoulomb = 1.2 ; short-range electrostatic cutoff (in nm)
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling
tcoupl = V-rescale ; modified Berendsen thermostat
tc-grps = Protein_JZ4 Water_and_ions ; two coupling groups - more accurate
tau_t = 0.1 0.1 ; time constant, in ps
ref_t = 300 300 ; reference temperature, one for each group, in K
; Pressure coupling
pcoupl = no ; no pressure coupling in NVT
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Dispersion correction is not used for proteins with the C36 additive FF
DispCorr = no
; Velocity generation
gen_vel = yes ; assign velocities from Maxwell distribution
gen_temp = 300 ; temperature for Maxwell distribution
gen_seed = -1 ; generate a random seed运行nvt模拟:
#生成执行文件tpr:
gmx grompp -f nvt.mdp -c em.gro -r em.gro -p topol.top -n index.ndx -o nvt.tpr#-n参数指定index文件,里面是
gmx mdrun -deffnm nvt4、进行压力耦合
下载官方的参数文件npt.mdp,注意修改里面的tc-grps参数,然后运行模拟:
gmx grompp -f npt.mdp -c nvt.gro -t nvt.cpt -r nvt.gro -p topol.top -n index.ndx -o npt.tpr
gmx mdrun -deffnm npt5、成品模拟:
经过温度耦合和压力耦合后,体系达到预期的室温和常压条件(旧制条件),即是温度在25°C,298.15K,压强在1.01bar,101.325kPa条件。
下载官方提供的md.mpt文件,可以进行成品模拟:
注意在这个md.mpt文件里是没有限制力参数的,没有下面这段参数:
define = -DPOSRES ; position restrain the protein and ligand这个参数是出现在nvt和npt模拟中,表示使用拓扑文件中的这个函数,对配体进行位置约束:
; Ligand position restraints
#ifdef POSRES #函数名是POSRES,mdp里就是DPOSRES,前面多个D
#include "posre_jz4.itp"
#endif另外,教程里的拓扑topol里还有水分子的位置限制参数,如下:
#ifdef POSRES_WATER
; Position restraint for each water oxygen
[ position_restraints ]
; i funct fcx fcy fcz1 1 1000 1000 1000
#endif但是mdp文件并没有define=-DPOSRES_WATER的参数。
运行没有约束的成品10ns模拟:
gmx grompp -f md.mdp -c npt.gro -t npt.cpt -p topol.top -n index.ndx -o md_0_10.tpr
gmx mdrun -deffnm md_0_10参考:
Protein-Ligand Complex (mdtutorials.com)
关于.mdp文件中的DPOSRES的问题 - 分子模拟 (Molecular Modeling) - 计算化学公社 (keinsci.com)
GROMACS中文手册:第三章 算法|Jerkwin
这篇关于Gromacs软件进行蛋白-配体体系MD模拟的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!






