本文主要是介绍【论文阅读】TomoTwin: generalized 3D localization of macromolecules in cryo-ET with structural data mining,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
题目
TomoTwin: generalized 3D localization of macromolecules in cryo-electron tomograms with structural data mining
利用结构数据挖掘对冷冻电子断层扫描中的大分子进行广义3D定位
发表期刊:Nature Methods
发表时间:2023.5.15
发表单位:德国多特蒙德马克斯普朗克分子生理研究所结构生物化学系
github 指路:https://github.com/MPI-Dortmund/tomotwin-cryoet/tree/main
论文指路:https://www.nature.com/articles/s41592-023-01878-z
摘要
冷冻电子断层成像技术使得对细胞环境进行高度详细地可视化成为可能。然而,仍然需要工具来分析这些密集体积中包含的全部信息。通过亚体积平均进行对大分子的详细分析需要首先在断层图中定位颗粒,这一任务受到多种因素的影响,包括低信噪比和拥挤的细胞空间。目前可用的方法在这方面要么容易出错,要么需要手动注释训练数据。为了帮助这一关键的颗粒拾取步骤,我们提出了TomoTwin:这是一个基于深度度量学习的冷冻电子断层图颗粒拾取的开源通用模型。通过在信息丰富、高维度空间中嵌入断层图,根据它们的三维结构将大分子分离,TomoTwin允许用户在断层图中自动识别蛋白质,无需手动创建训练数据或重新训练网络以定位新的蛋白质。
简介
冷冻电子断层成像技术(cryo-ET)已成为在其天然细胞环境中可视化大分子的重要技术,这使得在亚显微水平上观察到了多种重要结构。随着在低温下进行高压冷冻和聚焦离子束铣削等方面的进展,现在可以常规制备来自细胞甚至小型生物体的薄层(厚度小于200纳米)的样品。冷冻电子断层成像提供了在三维空间中以前所未有的细节捕捉细胞过程的独特机会,通过子体积平均(STA)对来自断层图的特定大分子的后续分析允许对原位大分子复合物进行深入的结构测定。特别是在结构预测的最新进展(如alphafold2)的补充下,STA构成了蛋白质生物化学和细胞蛋白质组学之间强大的桥梁。然而,要执行STA,首先必须在断层图中定位感兴趣的大分子的颗粒,这一任务由于这些数据的三维性质而变得复杂。
在亚显微水平上研究细胞生命的一个公认的障碍是在低温电子断层图中精确定位大分子。这导致了开发了几种基于深度学习的工具,通常利用流行的3D-Unet卷积神经网络(CNN)架构。然而,这些方法中没有一种能够展示泛化性,这意味着对于每个感兴趣的蛋白质,用户必须首先手动注释数百到数千个断层图中的粒子,并训练神经网络来识别该蛋白质。这不仅与自动断层图重建和STA的未来方向不兼容,而且对于大多数蛋白质组来说,从实验断层图中手动注释足够的训练数据以训练网络是不可能的。因此,这些深度学习工具目前无法帮助回答许多低温电子断层成像提出的巨大潜力所带来的未解生物学问题。
由于可用性的问题,模板匹配仍然在低温电子断层成像处理工作流中得到广泛应用,特别是那些强调吞吐量的工作流中,尽管与挑选准确性相比,这种技术在挑选准确性方面明显存在问题,通常限制了STA的整体有效性。
迄今为止,没有任何方法能够展示基于深度学习的挑选的准确性,并且在cryo-ET的可用性方面达到令人满意的水平。一种保持这种准确性的方法,同时规避对每个感兴趣的蛋白质手动注释训练数据的要求,是训练一个模型学习获得一种广义的3D分子形状表示,然后可以根据它们的结构区分大分子。这样的通用模型在二维(2D)单粒子低温电子显微镜(cryo-EM)分析的粒子挑选中得到了广泛应用,尽管将这些方法转化到断层图仍然存在问题,这是由于3D层析数据带来的额外挑战。
深度度量学习特别适用于这种具有挑战性的泛化情况,在这种情况下数据被编码为称为嵌入(embedding)的高维表示。在训练期间,模型会因为将来自不同类别的数据放在一起被惩罚,并且会因为将来自相同类别的数据放在嵌入空间中靠近的地方被奖励。因此在训练过程中,模型学会了在嵌入空间中将每个类别聚集在不同的区域,其中相似的类别更靠近,而不同的类别更远离。在某些情况下,数据集的嵌入被有序地排列,以允许在嵌入空间中对类别进行新的识别。通过理解相似性关系,深度度量学习模型已经展示了很强的泛化能力,能够根据它们与已知类别的相似性将新的数据类别放在嵌入空间中,而无需重新训练。
本文介绍了TomoTwin,一个用于结构数据挖掘的泛化颗粒拾取模型和深度度量学习工具包,适用于冷冻电子断层扫描图的研究。本文提供了两种使用TomoTwin进行大分子拾取的工作流程:一种是基于参考的工作流程,其中针对每个感兴趣的蛋白质选择一个单一分子并将其用作目标,另一种是从头的聚类工作流程,其中在二维流形上识别感兴趣的大分子结构。通过对多样化的模拟断层扫描图进行训练,TomoTwin的选取模型不仅可以在模拟数据中以高准确度定位新蛋白质,并且在实验断层扫描图中也能做到。通过去除训练数据标注和为每个蛋白质重新训练选取模型的步骤,TomoTwin 将基于深度学习的颗粒挑选的准确性与高度的易用性相结合,并允许在每个断层扫描图中同时选取多个感兴趣的蛋白质。
背后功能、构建和理念概述
TomoTwin的机器学习核心建立在学习断层扫描图中3D形状的广义表示原则上。通过深度度量学习进行训练,3D卷积神经网络(CNN)不仅能够定位来自训练集的大分子,而且也能泛化到新的大分子,使TomoTwin在避免为每个感兴趣的蛋白质重新训练的负担的同时保持深度学习颗粒挑选的高保真度。
通用模型通过对重叠的子体积进行采样并根据其大分子内容的相似性来对它们进行嵌入,以此来嵌入断层扫描图。嵌入断层扫描图后,可以通过识别嵌入空间中的相关区域来拾取每个大分子的颗粒。这可以通过识别断层图像中每种感兴趣蛋白质的单个示例并使用它们来标记它们嵌入的空间区域(基于参考的工作流程)来完成,或者通过将断层图像嵌入近似到二维流形来完成,其中每个大分子的簇可以通过肉眼识别(聚类工作流程)。一旦识别出包含感兴趣蛋白质的嵌入,就必须将它们映射回断层图像,其中同一分子的重叠选择可以合并为每个分子的一个集中选择。最后,TomoTwin允许用户根据每个感兴趣大分子的粒子大小以及每个粒子与嵌入空间中该大分子的目标嵌入之间的距离进行交互式筛选(图1d和Extended Data Fig. 1a)。
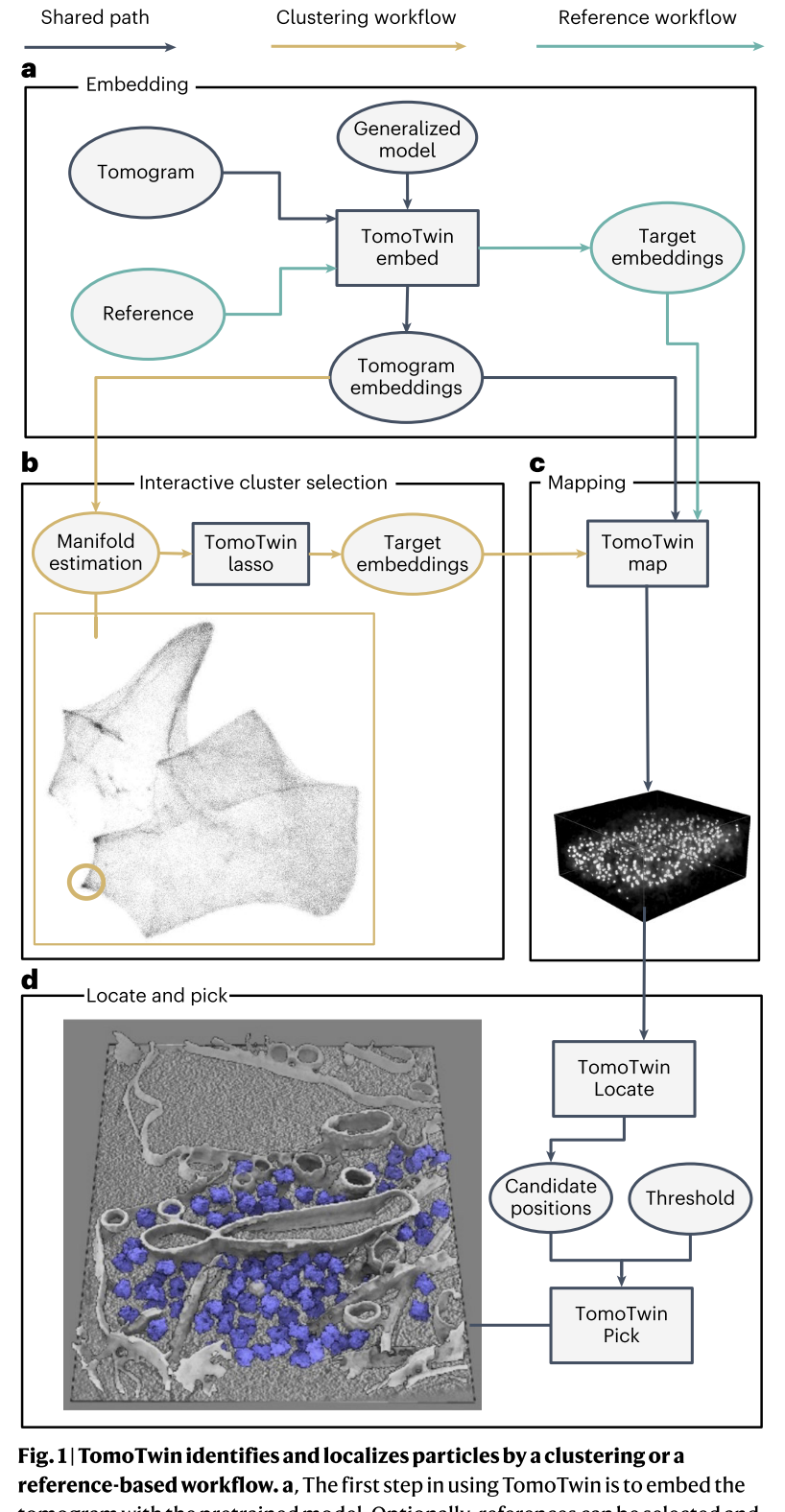
两个工作流程
TomoTwin 将断层图像嵌入高维空间,其中每个大分子的子体积位于空间中不同的区域。为了在嵌入空间中识别大分子的位置,我们提供了两种工作流程:基于参考的工作流程和聚类工作流程。每个工作流程都以高精度挑选粒子,但基于参考的方法始于在断层图中识别感兴趣蛋白质的示例,并将其映射到嵌入空间,而聚类工作流程则始于在嵌入空间中识别区域,并将其映射到断层图。对于任何给定应用程序,哪种工作流程最适用取决于在断层图与嵌入中蛋白质的识别容易程度。如果可以在断层图中识别感兴趣蛋白质的示例粒子,基于参考的工作流程提供了一种简化的挑选方法。相反,聚类工作流程对于探索断层图的大分子内容而无需先验知识提供了优势。两种工作流程共享TomoTwin嵌入功能的共同第一步,用于生成整个断层图体积的高维嵌入(图1a)。
在基于参考的工作流程中,将感兴趣蛋白质在断层图中的单个分子嵌入,生成该蛋白质的目标嵌入。在聚类工作流程中,断层图嵌入被近似到一个二维流形。然后可以直接使用这个二维流形使用交互式工具勾勒出一个或多个感兴趣的簇。然后将每个簇的封闭子体积的平均嵌入用作目标嵌入来代替参考。TomoTwin的Map功能以断层图嵌入和目标嵌入作为输入,并计算目标与嵌入中每个子体积之间的距离。这些距离被映射到子体积的位置,构建了在断层图中每个感兴趣蛋白质的建议粒子位置映射图(图1c)。Locate功能使用此映射来定位高相似性的峰值并生成候选粒子位置。最后,TomoTwin的Pick功能与图形用户界面一起使用这些候选位置,以及可调节的大小和相似性阈值,在断层图中挑选粒子,生成每个感兴趣蛋白质的坐标文件,适用于STA或其他分析(图1d)。
通用拾取模型的训练
TomoTwin的通用粒子挑选模型通过对来自模拟断层图的子体积三元组进行深度度量学习而进行训练。这些三元组构建为包含一个粒子的三个子体积的集合,其中两个包含相同的蛋白质,另一个包含不同的蛋白质,分别称为锚定(anchor)、正样本(positive)和负样本(negative)。使用从蛋白质数据银行(PDB)获取的一组120个在大小从30 kDa到2.7 mDa之间变化的结构不同的蛋白质来模拟84个断层图,其中包含总共120,000个粒子(扩展数据图2)。在训练期间,通过3D CNN嵌入子体积的批次将每个37×37×37子体积转换为位于32D高维嵌入流形上的32长度特征向量,该流形被塑造成32D高维球面的表面(扩展数据图1b)。然后,这些特征向量用于度量学习。
通过训练,TomoTwin学习将每个大分子放置在嵌入空间的一个明确区域,其中更相似的大分子彼此靠近,不相似的大分子则彼此远离(扩展数据图1d)。通过对大型、多样化的3D大分子形状和大小进行训练,TomoTwin学到了3D大分子形状的通用表示,并利用这一表示将新的大分子相对于其与已知蛋白质的结构相似性放置在嵌入空间中,而无需重新训练。
该挑选模型在蛋白质形状和大小上具有泛化能力
由于无法事先获取实验数据中所有分子在断层图中真实位置的信息,我们首先评估了训练模型在包含来自训练集的蛋白质的模拟断层图上的挑选性能。
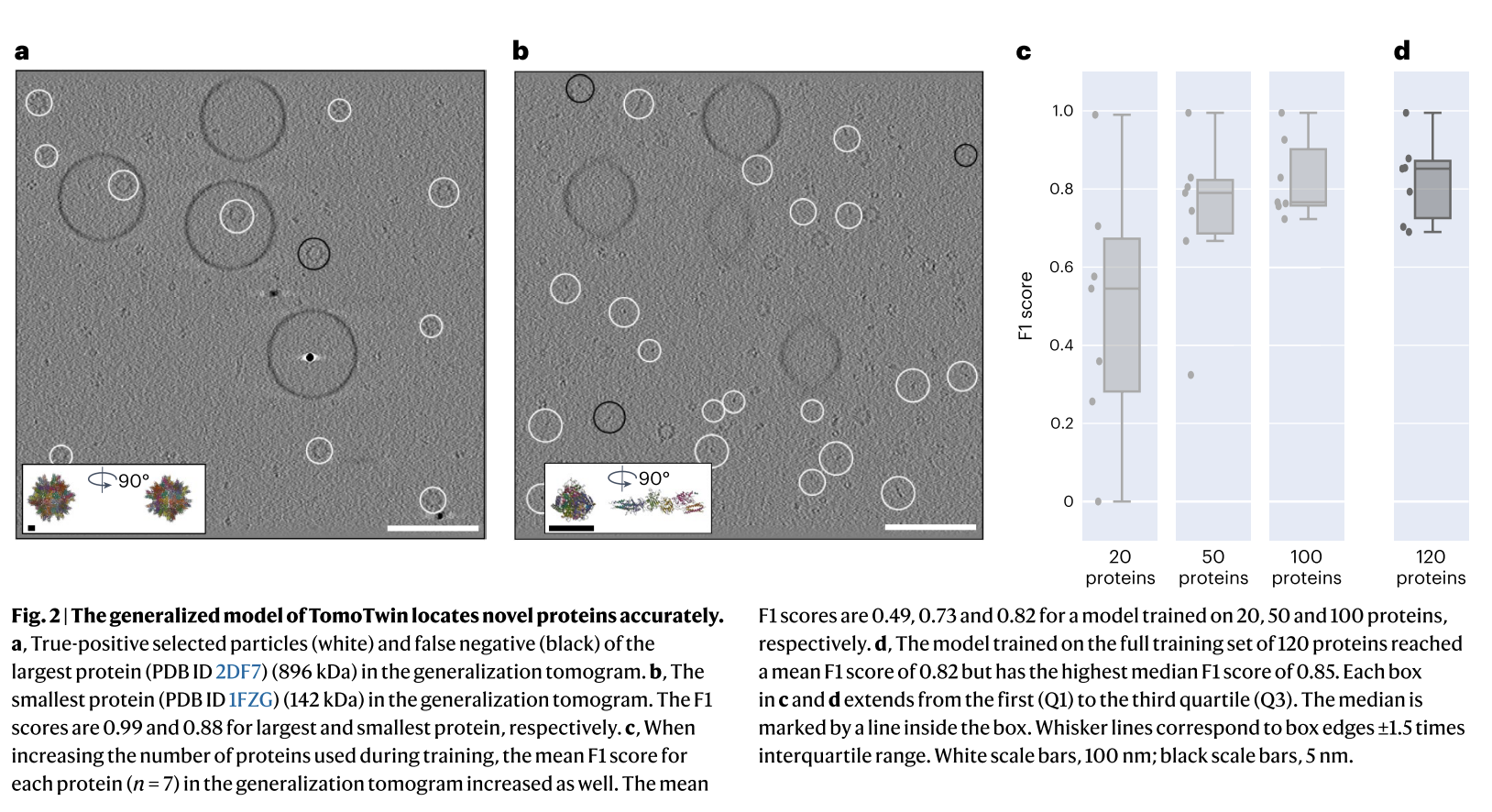
在所有验证断层图上的中位数F1挑选分数为0.88,范围为0.76到0.98(扩展数据图3a)。对于训练集中从30 kDa到2.7 mDa的所有蛋白质,中位数验证F1挑选分数为0.92(扩展数据图3b)。在罕见的情况下,观察到异常值分数,其中特定的蛋白质在一系列大小上无法被挑选(扩展数据图3c)。
对这些异常值的进一步检查表明,在模拟断层图中,与相似大小的蛋白质相比,每种蛋白质显示出特别弱的信号(扩展数据图3d)。在这些情况下,这些蛋白质在加权反投影的断层图重建过程中显示出无法很好恢复的形状。尽管如此,在验证断层图上的挑选表现显示出对各种形状和大小的蛋白质都具有很高的准确性。
TomoTwin对未知蛋白质的泛化性
为了评估通用挑选模型对未在训练数据集中的颗粒的泛化能力,我们使用基于参考的工作流测量了在模拟断层图上的挑选性能,该图包含训练集中未包含的蛋白质。当使用一组120种不同的蛋白质进行训练时(扩展数据图2),生成的模型能够在缺乏对这些蛋白质的先前训练的情况下,准确地定位所有七种蛋白质,中位F1分数为0.82(图2d)。为了量化训练集大小对泛化准确性的影响,我们对分别使用20、50、100和120种蛋白质进行训练的挑选模型进行了此分析,观察到随着训练集中蛋白质数量的增加,泛化准确性呈对数增加趋势(图2c)。这种在定位新型蛋白质方面的高准确性表明TomoTwin具有很高的泛化能力,这是迄今为止其他任何用于cryo-ET的深度学习挑选方法都没做到的。为了定量测量颗粒密度增加对挑选质量的影响,我们模拟了另一个泛化断层图,其中断层图中每种蛋白质的颗粒数量增加了五倍,以复制高度拥挤的环境(扩展数据图4a)。在这个密集的断层图中,我们观察到总体平均F1挑选分数为0.82,表明在包含许多蛋白质的密集环境中进行颗粒挑选带来了额外的挑战,但TomoTwin通用模型的挑选性能仍然是明确的。
TomoTwin在实验断层图中准确挑选蛋白质
由于TomoTwin完全是在模拟数据上训练的,因此研究其在实验断层图中挑选感兴趣蛋白质的能力至关重要。为了评估这一点,我们测试了TomoTwin在几个实验数据集上的挑选准确性。
首先,对包含三种蛋白质的混合样品进行了冷冻电子断层扫描,这三种蛋白质分别是apoferritin,RhsA,TcdA1,此外还包括脂质体(DOPC/POPC)(图3a)。
选择这种混合物是为了在体外创建一个复杂、拥挤的环境,可能会干扰挑选的准确性。使用通用模型为apoferritin、RhsA和TcdA1挑选了十个重建的断层图。在每种情况下,使用基于参考的工作流程,其中通过在一个断层图中挑选每种蛋白质的单个示例来创建目标嵌入。然后,将每种蛋白质的目标嵌入应用于数据集中的所有断层图进行挑选。通过直接可视化相似性图和挑选结果,TomoTwin在不受拥挤环境、污染物、降解颗粒或非目标蛋白质干扰的情况下,准确识别了混合物中的每种蛋白质,实现了高保真的挑选(图3b)。此外,还通过测量处于良好2D类别中的颗粒百分比来作为精度的相对指标(图3c和扩展数据图5c-e)。这通过手动计算TcdA1(召回率0.81,精度1.0)和apoferritin(召回率0.91,精度1.0)的召回率和精度(计数已挑选和未挑选的颗粒)来进一步确认(扩展数据图5a,b)。
这一挑选结果证实了TomoTwin在断层图中进行颗粒挑选的多个方面的泛化能力。首先,TomoTwin可以推广到新的蛋白质,而无需重新训练,因为这些蛋白质中没有一个包含在训练集中。同样重要的是,高度保真的挑选结果表明,经过模拟断层图训练的TomoTwin也可以应用于实验断层图。

冷冻电子断层图技术的主要优势之一是能够直接在细胞自然环境中可视化蛋白质。然而,由于细胞空间的拥挤和厚样本引起的对比度不足,蛋白质在细胞环境中的定位存在相当大的挑战。为了评估其在细胞断层图中定位颗粒的能力,我们将TomoTwin应用于包含肺炎支原体的断层图数据集[EMPIAR 10499]。使用TomoTwin通用模型,我们使用基于参考的工作流,在65个断层图中挑选了70S核糖体,其中在一个断层图中确定了一个单一的核糖体,并用于生成一个目标嵌入,然后应用于挑选整个数据集。为了可视化结果,我们提取了伪亚断层图,并使用一个70S核糖体冷冻电子显微结构(EMD-11650)进行了3D分类,低通滤波到30埃作为参考。由于所有的3D类别都类似于大约15埃的精修核糖体,这清楚地表明TomoTwin在细胞断层图中也能够进行高度准确的挑选。
TomoTwin建立了准确性和可用性的新标准
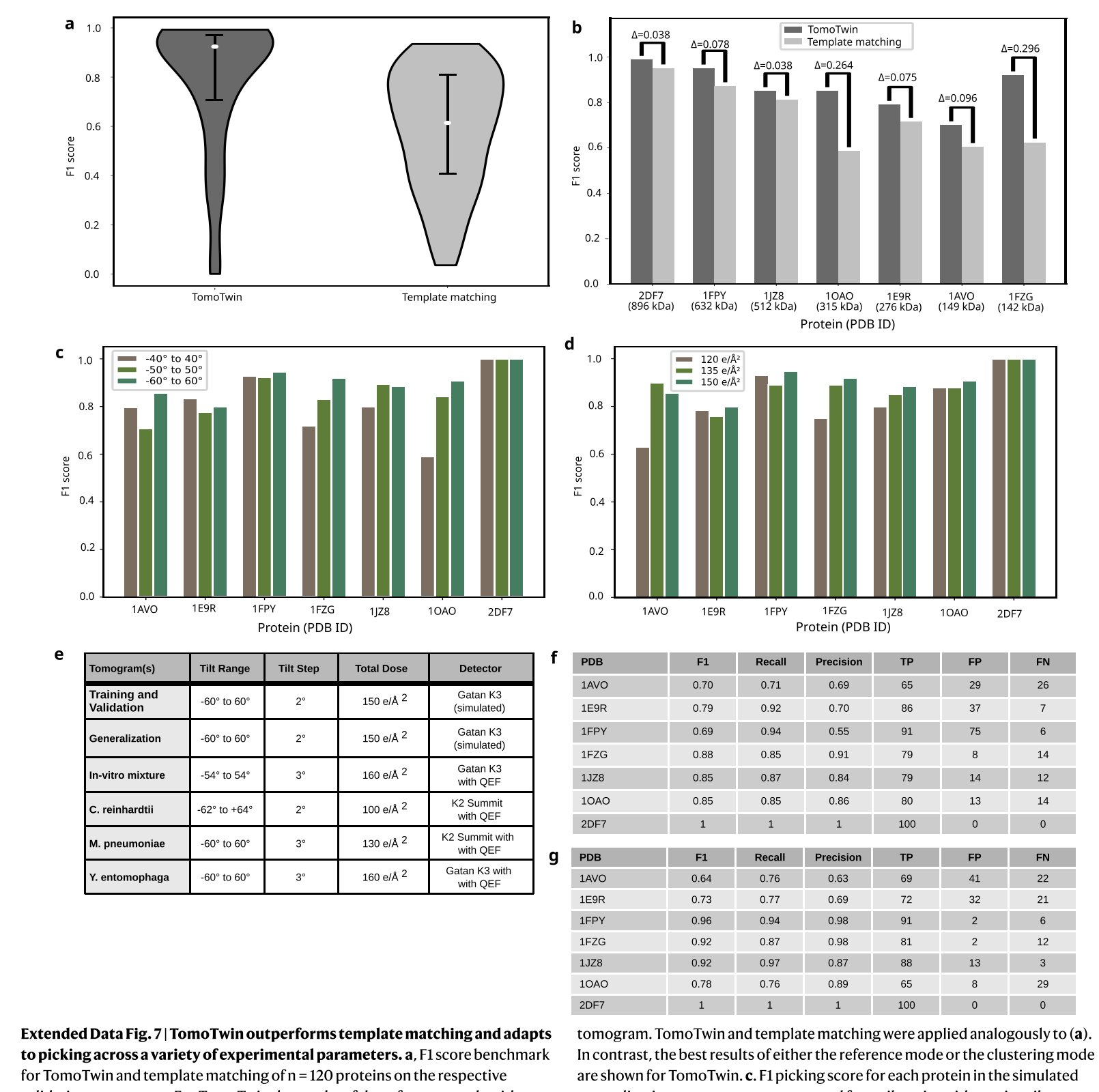
为了确定TomoTwin与cryo-ET颗粒挑选的现有方法相比如何,我们首先直接测量了其挑选性能,首先与模板匹配比较,然后再与非泛化机器学习工作流比较。
在cryo-ET的模板匹配中有几种可用的软件,包括EMAN2、Dynamo和PyTom,我们选择了EMAN2作为代表。当应用于包含30 kDa到2.7 mDa大小范围的120种蛋白质的验证数据集时,TomoTwin表现出了优越的挑选性能,通过F1准确度分数的测量以及在整个蛋白质范围内挑选准确性的更大一致性来衡量(扩展数据图7a)。此外,这一优势也体现在泛化断层图的挑选性能上(扩展数据图7b)。
与非泛化机器学习挑选工作流直接比较TomoTwin更具挑战性,因为主要区别在于可用性而不是统计挑选精度。非泛化深度学习方法,如DeepFinder,相对于模板匹配的优势已经在使用比模板匹配更少的粒子实现对酶核糖-1,5-二磷酸羧化酶氧化酶(RuBisCO)进行低分辨率重建时得到证实。然而,为了实现这一目标,首先使用模板匹配在 tomograms 中挑选 RuBisCO,然后手动筛选挑选结果,以获取超过 175,000 个注释粒子,以训练和验证在其余 tomograms 中挑选该酶的模型。尽管总体上产生了很高的准确性,但在这个工作流程中仅训练步骤就需要 35 小时。这个样本特别适用于评估在拥挤的细胞环境中进行粒子挑选的性能,因为 RuBisCO 会进入堆积在 pyrenoid 内部的密集蛋白基质中,这是一种不同寻常的现象。在应用于相同数据集的 tomogram(EMPIAR 10694)时,TomoTwin 能够使用单一参考物来定位 RuBisCO,召回率为 0.8(Extended Data Fig. 8)。这些挑选结果被证明足以通过 STA 进行直接分析,超过了先前报道的分辨率,同时保持了高效的工作流程(Fig. 4)。通过比较 TomoTwin 原始挑选粒子的 RuBisCO 地图与原始地图(EMD-3694)42,可以看出使用 TomoTwin 挑选的粒子进行的重建在质量上是可比较的,如果不是更高的质量,因此 TomoTwin 挑选不限制 STA 可以实现的分辨率(Extended Data Fig. 9e)。此外,由于不需要用户手动注释训练数据或重新训练网络来挑选这些粒子,TomoTwin 能够在总工作时间不到 1 小时的情况下生成这些准确的挑选坐标。在应用于相同的 tomogram 时,聚类工作流实现了类似的挑选结果(Extended Data Fig. 8)。
TomoTwin 在各种实验设置中具有泛化性
除了泛化以挑选新蛋白质外,高度可用的挑选工具还必须泛化于冷冻电子断层扫描中的各种常见实验设置。
尽管仅对总剂量为 150 e/Å2 的模拟断层图上进行训练(从 -60° 到 60° 的倾斜序列,以 2° 为增量收集),但在实验断层图上进行的上述挑选示例展示了几个额外的泛化程度,包括倾斜范围、倾斜步长、总剂量和探测器(Extended Data Fig. 7e)。
为了进一步定量分析不同实验参数的影响,创建了几个一致的模拟断层图,其中倾斜范围和总电子剂量发生变化,以考虑一系列可能的冷冻电子断层实验设置(Extended Data Fig. 7)。为了控制用于挑选的参考颗粒可能存在的偏差,每种蛋白质使用了五个参考颗粒,并选用了在参数中返回最一致的挑选结果的那个参考。
减小倾斜范围会增加缺失楔伪影对重建断层图像的影响,从而导致每个重建颗粒的变形更强。这种效应对于一氧化碳脱氢酶等长而细的颗粒尤其明显,使得精确的颗粒拾取变得更具挑战性。总体而言,当倾斜范围限制在-60°60°到-50°50°和-40°40°时,我们观察到平均F1拾取性能分别下降5.4%和10.3%。同样,减少总电子剂量会直接减少蛋白质信号,从而导致断层图像的信噪比降低。我们观察到当总剂量限制在150至135和120 e/Å2时,F1拾取性能分别下降2.3和8.6%。
总体而言,虽然预计拾取性能会随着断层图像中蛋白质信号的破坏或缺失而下降,但TomoTwin利用基于其深度度量学习猪肝的高度适应性来实现精确的颗粒拾取。
嵌入流形上的结构数据挖掘
TomoTwin 的嵌入功能将断层图映射为根据其大分子内容组织的一系列嵌入。这种有组织的嵌入为结构数据挖掘提供了理想的选择。这些嵌入可以通过二维流形上的近似直接可视化(图5A,C)。通常,这些二维图中包含一个大的细长块,对应于背景嵌入和整体降维效果的组合,以及其他明确定义的簇。这些簇代表了 tomogram 中的常见结构基元,包括突出表达的蛋白质,以及在实验数据的情况下,膜、基准物和包含偏离中心蛋白质的嵌入。TomoTwin的聚类工作流允许用户交互式地识别感兴趣的簇,并生成粒子挑选的目标,而无需任何先验知识关于 tomogram 内容。然而,它确实要求蛋白质的丰度足够高以形成簇。
为了定量评估基于聚类的挑选准确性,我们评估了使用聚类挑选时对于广义断层图中每个蛋白质的 F1 挑选分数(图 5b、e 和扩展数据图 7f、g)。基于聚类的拾取以高精度识别了各种大小范围内的每种蛋白质,表明这种工作流提供了一种替代但有效的粒子挑选方法。在可视化嵌入中还值得注意的是,各个蛋白质簇是按照大小在整体上组织的,这表明该模型将大分子形状的相似性表示为嵌入空间中的距离(图 5a)。
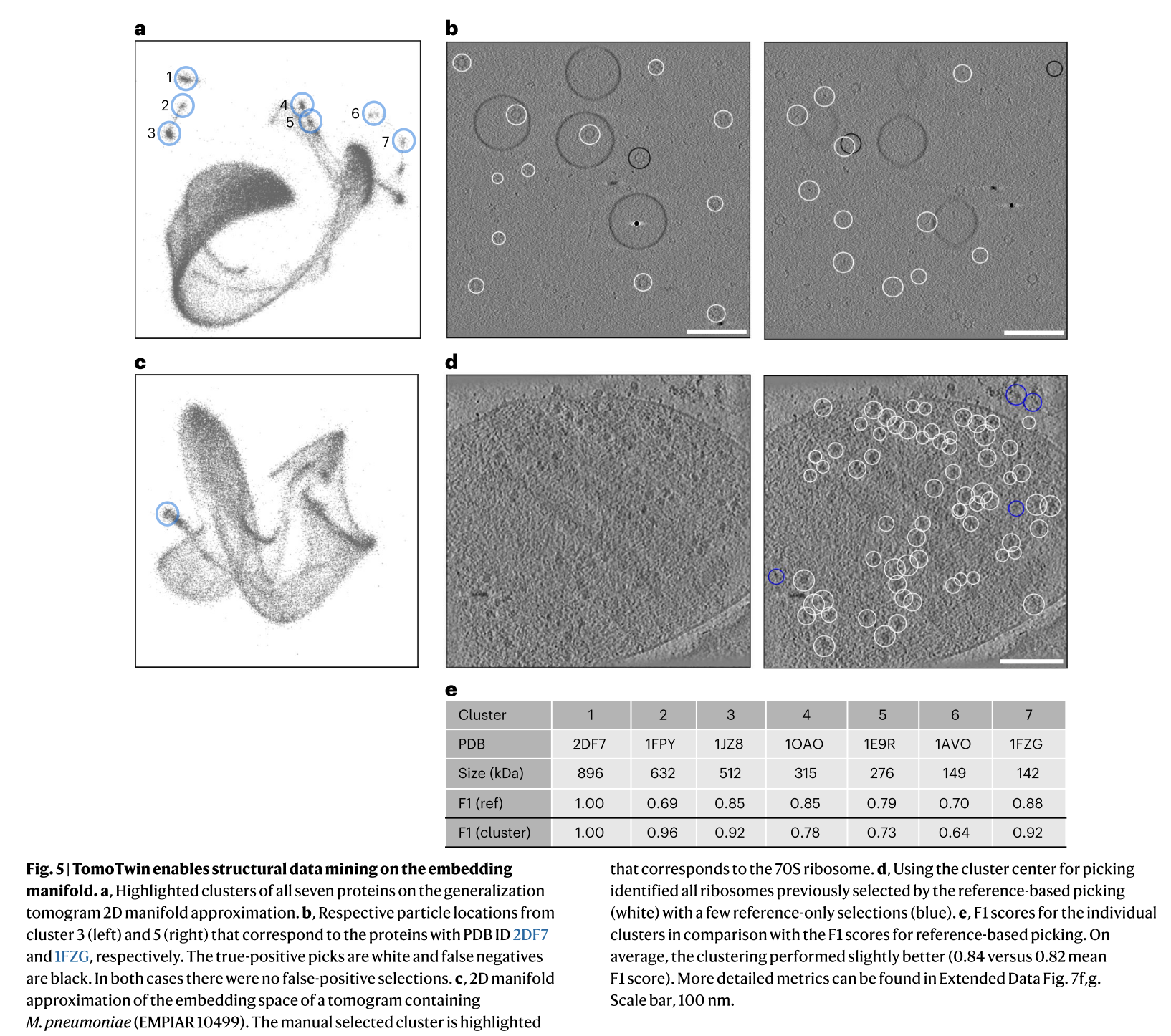
我们还针对含有肺炎支原体的细胞断层图,将基于聚类的挑选与基于参考的方法进行了比较(图5 C)。可视化这些断层图像的嵌入可以看到几个簇。其中一个在选取后可以产生70S核糖体的准确颗粒位置,与基于参考的方法产生的颗粒位置几乎相同,进一步强调了两种工作流程的鲁棒性。
最后,关于通过 cryo-ET 对原位大分子进行研究的一个突出问题是,通过 STA 可以合理研究多少蛋白质组。毫无疑问,通过去噪、缺失楔修复或其他方法改善整体断层图像对比度将会产生重大影响,但用于从头蛋白质定位的 TomoTwin 的聚类工作流代表了一种迄今为止未被记录的通过 cryo-ET 探索蛋白质组区域的方法。为了探索原位聚类工作流程的应用范围,我们将其应用于包含经过受控细胞裂解的 Yersinia entomophaga 的断层扫描。嵌入断层图像后,对一张断层图像执行聚类工作流程显示了多个聚类簇,其中两个对应于断层图像中可识别的生物分子(图 6a、b)。提取每个簇的拾取坐标并使用球形参考进行 STA,以评估它们包含的生物分子(图 6c)。对第一个簇的细化(15.5 Å)很容易发现该簇包含 70S 细菌核糖体(图 6d)。第二个簇包含一个相当小的生物分子。通过球形参考对这些选取进行 STA 的细化得到了一个低分辨率(18.6 Å)重建,显示了一种未经 STA 描述的蛋白质,其形状与细菌 RNA 聚合酶大致相同(图 6d)。已知RNA 聚合酶以高丰度表达,尽管在这种分辨率下,通过细化直接鉴定蛋白质并不绝对。值得注意的是,TomoTwin 将同一蛋白质的颗粒聚集在一起,无论它们是否保持在细胞内部或由于裂解在邻近细胞,这表明蛋白质的嵌入在各种周围环境中都保持一致。
讨论
尽管提供了在天然细胞环境中研究蛋白质的潜力,但目前只有少数蛋白质通过冷冻电子断层和STA成功研究。在某种程度上,这是因为随着细胞环境的增加、大分子复合物的行程以及较厚样本导致的对比度较差,为后续STA挑选单个蛋白质带来了挑战。为了协助这一关键的颗粒拾取步骤,我们开发了TomoTwin,这是一种基于深度度量学习的强大的冷冻电子断层扫描通用拾取模型。TomoTwin允许用户从头识别断层图像中的蛋白质,而无需在每次定位新蛋白质时手动创建训练数据或重新训练网络。
TomoTwin提供了两种互补的颗粒拾取工作流程。基于参考的工作流程用于在断层图中挑选易于观察的大分子,而聚类工作流程则提供了一个独特的机会,无需先验即可交互式探索断层图内容。虽然目前其拾取效用收到断层图像重建过程中可实现的整体对比度的限制,但聚类工作流程与断层扫描重建的未来进展相结合,提出了一种拓展STA可以访问的蛋白质组比例的潜在方法。
冷冻电子断层中的缺失楔信息会导致重建颗粒的变形,特别是沿xz平面的变形。然而,在某些生物学背景下,准确定位感兴趣的蛋白质在这个视图中变得非常重要。与将图谱视为一系列 xy 视图的典型观点相反,TomoTwin 直接在 3D 中选择粒子,因此不仅在 xy 平面上,而且沿 z 轴方向也能实现准确的选择。这使用户能够自信地沿其选择的轴查看选择,并选择在仅从 xy 平面查看时可能难以发现的粒子(扩展数据图 4b–f)。

此外,对于提高数据吞吐量的需求需要在 cryo-ET 中开发自动处理算法。TomoTwin 可以轻松集成高吞吐量的断层扫描重建和 STA 工作流,并且当与无监督聚类算法结合使用时,TomoTwin 为整个断层扫描级别上的无监督STA 分析铺平了道路(扩展数据图 9)。
虽然 TomoTwin 的粒子拾取在准确性和易用性方面具有优势,但它并非没有局限性。目前,该模型不适用于拾取膜蛋白或纤维。此外,在聚类工作流中,蛋白簇的出现在很大程度上取决于断层图像中蛋白质的拷贝数。例如,在 Y. entomophaga 细胞中,仅观察到七个 Tc 毒素分子,使得该蛋白在聚类工作流中无法识别,尽管在基于参考的工作流中可以迅速而准确地拾取。此外,因为 TomoTwin 是以 10 Å 的像素大小训练的,所以当前模型不设计在粒子拾取级别区分同一蛋白的多个构象。最后,尽管训练数据包含小至 30 kDa 的蛋白,但在普遍适用性阶段,准确拾取的预期下限约为 150 kDa,这是因为 TomoTwin 在缩小的断层图像上进行拾取(每像素约 10 Å)。虽然比这个大小更小的粒子理论上可以在实验断层图像中定位,但通过 STA 评估这些拾取的准确性目前还超出了该领域的限制。
TomoTwin 是一个稳健的、开源的工具,用于在冷冻电子断层扫描图中定位颗粒。它充分利用深度学习的优势,同时保持了高度的可用性,使其能够在各种冷冻电子断层扫描实验中发挥作用。TomoTwin 的开发和训练代码以及通用拾取模型可在 https://github.com/MPI-Dortmund/tomotwin-cryoet 获取。
方法
训练数据生成
TomoTwin是在123个数据类别上进行训练的,这些类别由来自模拟扫描图的120种不同蛋白质、膜、噪声和基准点的子体积组成。为了确保TomoTwin在尽可能多样化的蛋白质集合上进行训练,从PDB中选择了108种大小从30kDa到2.7mDa的蛋白质,并计算了每种蛋白质的10埃低通滤波图之间的互相关性(Extended Data Fig. 2)。任何与训练集中的另一种蛋白质具有高度相似性(大于0.6)的蛋白质都被标记为替换。此外,还包括了2021年SHREC竞赛的数据,其中包含12种蛋白质,总共训练了120种蛋白质。通过在训练集中为每个数据类别提供800个子体积和在验证集中提供200个子体积,实现了训练/验证的分割,总训练集大小为98,400个子体积,验证集大小为24,600个子体积。
断层扫描模拟
使用TEM Simulator进行断层扫描模拟,计算单个蛋白质的散射势并将它们放置在体积内的可定义位置。模拟的输出是一个倾斜系列,然后使用IMOD进行重建。生成了一个配置文件,其中包含有关电子束、显微镜光学器件、探测器、倾斜几何形状和样品体积的属性。默认探测器经过调整以反映现代Gatan K3相机的MTF曲线,其量子效率为0.9。将探测器大小设置为1,024×1,024,像素大小为5微米。将放大倍数设置为9,800,球差和色差分别调整为2.7和2毫米,以模仿流行的现代透射电子显微镜(TEM)。选择了一个80微米的照明器光圈。对于每个层析图,偏焦值在-2.5到-5微米之间随机选择。使用-60°至+60°的2°步长的倾斜方案。为了简化和优化模拟过程,我们编写了一套名为“tem-simulator-scripts”的开源程序(https://github.com/MPI-Dortmund/tem-simulator-scripts)。这些脚本需要PDB文件作为输入进行模拟,并指定每个PDB要模拟的粒子数。然后,该程序使用以下流程生成了用于本研究的重建的层析图:
(1)在体积内生成密集排列的随机粒子位置,确保各个粒子之间不重叠。
(2)生成occupancy map:一个体积,其中每个体素根据蛋白质的身份进行标记。
(3)生成基准点图 fiducial maps。
(4)生成囊泡图 vesicle maps。
(5)为TEM-simulator生成配置文件。
(6)使用TEM-simulator进行倾斜系列的模拟。
(7)使用IMOD进行对齐和重建。
上面的步骤也可以单独执行,来完全控制所有参数。
使用这个程序,我们模拟了11组蛋白质。这些组一共包含了108种不同的蛋白质,每组涵盖不同大小的蛋白质。对于每组,我们模拟了8张断层图,尺寸为512512200体素,像素大小为1.02nm,和不同的蛋白质密度。对于断层图1、2和8,每个蛋白质生成150个颗粒,对于断层图3和4,每个蛋白质生成125个颗粒,对于5和6每个蛋白质生成100个颗粒,对于7,每个蛋白质生成75个颗粒。断层图1-7用于训练,断层图8用于验证。本研究中使用的生成的断层图以及所有元数据都是公开的。这些模拟数据用于构建训练和验证集,以评估网络训练、颗粒定位和模型泛化性。
卷积网络结构
为了将体积性的冷冻电子断层扫描(cryo-ET)数据编码为高维空间中的嵌入向量,TomoTwin 使用了一个由五个卷积块组成的3D卷积神经网络(CNN),后跟一个头网络(Extended Data Fig. 1b)。每个卷积块包括两个核大小为3×3×3的3D卷积层。每个卷积层后面跟着一个归一化层和一个Leaky ReLU 激活函数。在每个卷积块的第一个卷积层中,输出通道的数量是输入通道的两倍,而在第二个卷积层中,输出通道的数量与前一层的输出相匹配。在第一个卷积块之后执行2×2×2的最大池化,最后一个卷积块之后执行自适应最大池化,将大小调整为2×2×2。因此,当提供一个37×37×37的子体积,其中通道数为1,作为一个归一化的37×37×37×1的数组时,卷积块将输入转换为一个2×2×2×1,024的特征向量,然后将其馈送到头网络。在头网络中,首先将特征向量以通道为基准展平,然后通过一个丢弃层,最后通过一系列全连接层将展平的向量转换为32D特征向量。最后,这个特征向量进行L2归一化,生成一个子体积的输出嵌入向量。
三元组生成
TomoTwin 是在由锚定体积 A、正体积 P 和负体积 N 组成的三元组子体积上进行训练的(Extended Data Fig. 1c)。每个子体积都分配给一个与其中的大分子相对应的数据类,并且大小为 37×37×37 个体素。构建三元组,其中 A 和 P 从相同的数据类中采样,而 N 从不同的数据类中采样。给定一个距离函数 D 和一个嵌入函数 f,三元组损失被定义为:
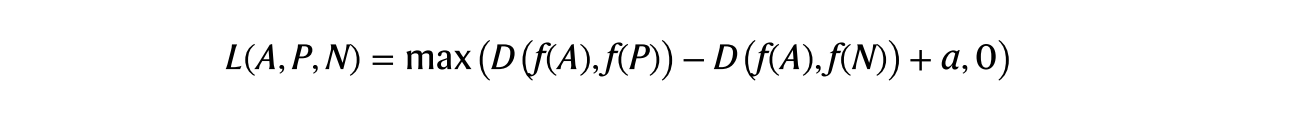
其中超参数α是边距值。作为距离函数D,我们使用余弦相似度,定义为:
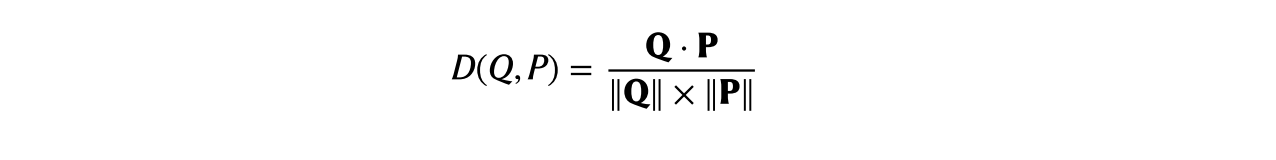
其中 Q 和 P 是任意嵌入向量,• 是点积,‖⋅‖ 是向量的长度。在训练期间,通过在线半硬三元组挖掘生成三元组,其中一批子体积被嵌入,并自动生成三元组,负子体积嵌入的选择仅限于与锚定体积的距离大于正子体积嵌入但不大于 α m i n e r α_{miner} αminer 边距的那些子体积:
D ( a , p ) < D ( a , n ) < D ( a , p ) + α m i n e r D(a,p)<D(a,n)<D(a,p)+\alpha_{miner} D(a,p)<D(a,n)<D(a,p)+αminer
其中a/p/n分别是anchor,positive和negative对应的嵌入向量。
通用拾取模型训练
使用ADAM优化器对3D CNN进行了600epoch的训练。来自在验证集中对子体积进行最佳 F1 分数评估的时期的模型进一步在本地化和泛化任务中进行评估,并用作通用的拾取模型。
数据增强
在计算嵌入之前,对每个标准化体积应用在线数据增强,包括旋转、丢弃、平移和添加噪声。对于旋转增强,子体积在xy平面上旋转随机角度,但不是xz和yz,以防止缺失楔重新定向。在丢弃增强中,随机选择 5% 到 20% 的体积元素,将其设置为子体积的均值。在平移增强中,将子体积在每个方向上平移 1-2 个像素。添加噪声增强会向子体积添加标准差在 0 到 0.3 之间的随机选择的高斯噪声。
超参数优化
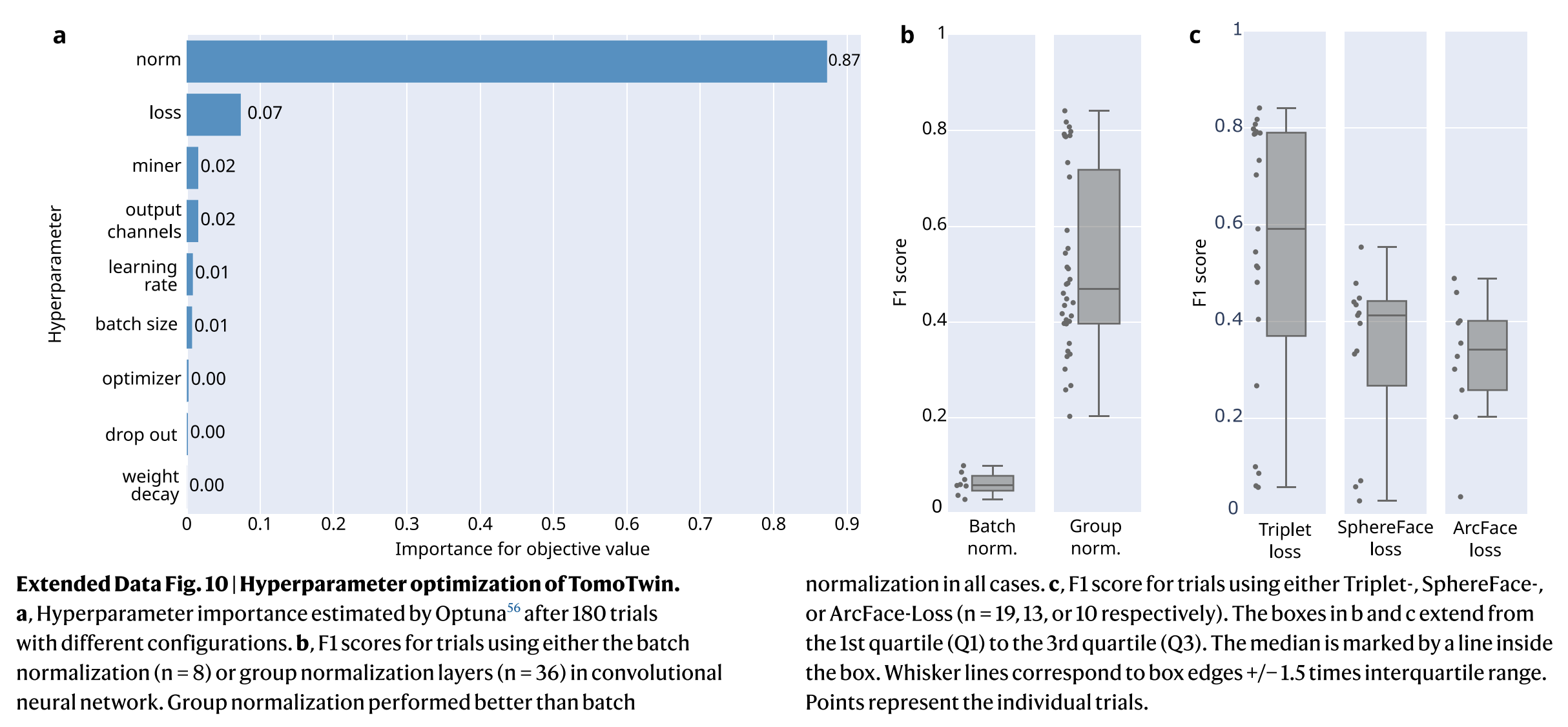
现代 CNN 的训练涉及许多超参数的选择,其中一些选择影响架构,而另一些影响学习过程本身。虽然存在一些启发式方法来指导超参数的选择,但通过手工找到最大化机器学习工具效用的设置组合很快变得棘手。我们使用 Optuna 探索超参数搜索空间,找到了一组优化的训练参数。模型在训练数据的子集上进行了 200 轮的训练,并在每轮之后计算了验证集上的 F1 分数。对于训练运行中 F1 分数低于全局中位数的情况,进行了 50 轮后修剪。总体上,我们对学习率、丢失率、优化器、批量大小、权重衰减、第一个卷积核的大小、输出层节点数、在线三元组挖掘策略(semihard,easyhard,none)、归一化类型(group norm,batch norm)、损失函数(TripletLoss,Sphere-Face,ArcFace)和损失边际进行了搜索(详见扩展数据图 10)。
值得注意的是,训练过程中应用的归一化类型是性能的最大整体影响因素,其中 group normalization 优于更常见的批归一化策略(详见扩展数据图 10b)。此外,值得注意的是标准三元组损失函数在性能上优于理论上更优越的 SphereFace 和 ArcFace 损失函数(详见扩展数据图 10c)。这些发现强调了在训练期间探索各种超参数的必要性,因为仅靠启发式方法无法足够指导现代 CNN 训练的最佳超参数选择。
使用通用模型的颗粒拾取工作流程
对于使用通用模型拾取的每个数据集,首先对所有断层体进行嵌入。为此,将tomograms细分为一系列重叠的 37 × 37 × 37 子体积,步幅为 2 个体素。对于基于参考的工作流程,选择每个感兴趣的蛋白质的一个随机颗粒作为参考并进行嵌入,以生成目标嵌入。将tomogram和target嵌入提供给 TomoTwin Map,该工具计算了每个目标嵌入与来自断层体每个子体积嵌入之间的距离矩阵,并返回了每个目标嵌入的相似性映射。然后将此矩阵提供给 TomoTwin Locate,该工具使用基于区域生长的最大检测过程识别高置信度区域,然后进行非极大值抑制。然后,在 TomoTwin 图形用户界面中,对返回的候选位置进行置信度和大小阈值处理,以生成每个感兴趣蛋白质的最终坐标。
模拟数据验证
粒子定位性能通过三个度量值进行计算:召回率(recall)、精确度(precision)以及两者的谐波均值 F1 分数。
训练模型的粒子定位准确性是通过对验证集中每个层析图进行评估的(Extended Data Fig. 3a)。为了测试模型的泛化性能,进行了包含七种在训练集中未包含的蛋白质的层析图的定位任务,因此TomoTwin在这方面是初始的(Fig. 2)。
聚类
为了进行聚类分析,将来自高维断层图像嵌入的40万个嵌入的随机样本拟合到具有均匀流形近似(UMAP)的均匀二维流形,并使用RAPIDS包提供的GPU加速。
UMAP模型被用作转换整个断层图嵌入的基础,并绘制了结果(图5a,c)。通过眼睛识别并通过绘制一个包含所需点的封闭形状来选择簇。然后,将封闭的点追溯回它们的原始高维嵌入,并计算它们的平均嵌入。然后将该平均嵌入用作分类、定位和拾取的目标嵌入,与基于参考的工作流程相同。
实验数据验证
实验数据无法获得真实颗粒坐标,使得精度和召回率等性能指标的计算变得复杂。估计这些的一种方法是手动计算完整断层图像或参考区域的真阳性、假阳性和假阴性。
然而,由于由于手动拾取某些蛋白质的困难,这种方法并不总是可行,另一种测量不引入参考偏差的拾取精度的方法是在每个蛋白质的选定坐标处提取子体积,使用SPHIRE将3D子体积投影到2D,并进行无参考的2D分类。对少量粒子进行2D分类可能会导致不相关的结果,因为只有蛋白质最常见的姿势才会有足够的颗粒进行有效聚类,可能导致许多真阳性粒子被错误地分类为假阳性。尽管如此,仍测量了良好的2D类别中的颗粒百分比作为精度的指标。
对于在实验室准备的样本的样品断层图,使用TomoTwin识别的颗粒坐标被缩放至5.936Å每像素,以匹配最初重建的断层图。将这些断层图导入,并使用这些坐标在Relion v.3.0 中提取子断层图。对于无参考分析,3D子层析图通过SPHIRE投影到2D,然后用于2D分类。
获得包含Mycoplasma pneumoniae的层析图来自EMPIAR 10499,使用TomoTwin识别的颗粒的坐标进行了缩放,以匹配最初重建的层析图像素大小为6.802 Å每像素。将这些层析图导入,导入坐标并用于在Relion 4.0中重建伪子层析图。从一个70S核糖体(EMD-11650)中通过低通滤波到30 Å并将像素大小缩放到6.802 Å每像素来创建参考。使用这个参考与Relion v.4.0中的伪子层析图进行3D分类。
包含Chlamydomonas reinhardtii细胞的层析图的倾斜系列和对准文件来自EMPIAR 10694,用于以每像素13.68 Å的像素大小进行颗粒挑选,以及每像素6.84 Å的像素大小进行STA的层析图重建。使用TomoTwin基于参考的工作流程进行RuBisCO的挑选,然后将结果的坐标缩放到像素大小以进行提取,并随后在Relion v.3.0中进行STA。提取的子层析图用于使用通过将RuBisCO的已知模型(PDB 1BXN)低通滤波到60 Å生成的参考进行初始3D细化。该图用于拟合RuBisCO模型。
重建的断层图像素大小缩小到9.288 Å每像素,并使用TomoTwin聚类工作流程进行挑选。每个聚类的挑选坐标被重新缩放到4.644 Å每像素以进行提取。将这些断层图导入,并使用这些坐标在Relion v.3.0中提取子断层图。使用一个球形参考进行初始3D细化,以消除参考偏差的可能性,并使用得到的map进行无对齐的3D分类。最后,使用3D类别之一作为参考重复进行3D细化,以获得一张一致的3D细化图,然后用于拟合候选蛋白质的PDB模型。
硬件
计算过程中使用了两个计算机设置,一个是分布式计算系统,一个是本地工作站。分布式计算系统包括由最大普朗克学会超级计算机“Raven”组成,最多可使用30个Nvidia A100图形处理单元(GPU),每个GPU具有40 GB内存。每个进程使用Intel Xeon IceLake-SP 8360Y处理器的18个内核和128 GB系统内存。本地工作站包括一个本地单元,配备有Nvidia Titan V(12 GB内存)GPU和Intel i9-7920X CPU,64 GB系统内存。
Hyperparameter optimization was done in parallel for 7 d once
the distributed computing setup and embeddings were calculated on
this, also using two GPUs(注:这里没理解)。在所有情况下,嵌入使用了37的包围盒大小和2的步幅。本地工作站用于各种任务,并使用两个GPU计算时间。
用时
计算嵌入是TomoTwin需要大量处理时间的唯一功能。为了测量这一点,我们在本地工作站和分布式计算系统上嵌入了我们最大的实验断层图(Fourier缩小后为608×855×148)。使用两个GPU,在本地设置上断层图像嵌入需要80分钟,在分布式设置上需要30分钟,这对应于在每个设置上挑选每个断层图中所有感兴趣的蛋白质所需的总时间。
统计和重现性
由通用模型生成的嵌入是确定性的,允许用户随时重现挑选结果。在用于聚类工作流程中的可视化的Nvidia RAPIDS UMAP是唯一不确定性的包。然而,由于挑选和阈值计算始终在原始嵌入上执行,因此整体挑选结果保持一致。
A u t h o r : C h i e r Author: Chier Author:Chier
这篇关于【论文阅读】TomoTwin: generalized 3D localization of macromolecules in cryo-ET with structural data mining的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!