本文主要是介绍DRAM(Distilling and Refining Annotations of Metabolism,提取和精练代谢注释)工具安装和使用,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
先看文章介绍吧:DRAM for distilling microbial metabolism to automate the curation of microbiome function | Nucleic Acids Research | Oxford Academic (oup.com)
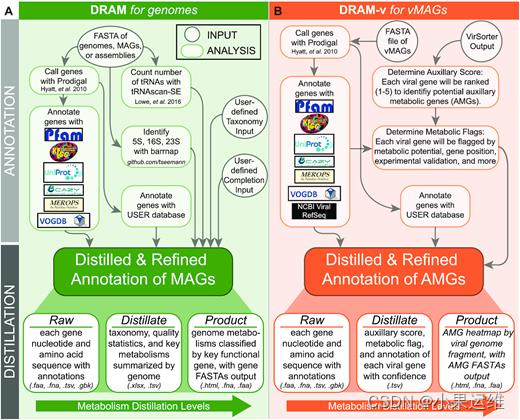
1、安装
默认使用conda安装吧,也建议使用conda,pip安装其实都差不多,但容易破坏当前环境。
###下载环境依赖列表
wget https://raw.githubusercontent.com/shafferm/DRAM/master/environment.yaml###使用环境依赖文件创建DRAM的conda环境
conda env create -f environment.yaml -n DRAM###激活DRAM环境
conda activate DRAM
#有的环境可能还是source
source activate DRAM附gitub仓库安装说明地址,如果打不开的就直接使用上面的命令安装吧:
2. How to Install and Set Up DRAM · WrightonLabCSU/DRAM Wiki (github.com)
2、配置数据库
先查看DRAM的配置情况:
###激活DRAM环境后,使用print_config命令查看当前数据库配置
DRAM-setup.py print_config
##很显然,这里数据库都还没有配置好,报错了。
2023-11-23 16:00:45,883 - Logging to console
/anaconda3/envs/DRAM/lib/python3.10/site-packages/mag_annotator/database_handler.py:123: UserWarning: Database does not exist at path Nonewarnings.warn("Database does not exist at path %s" % description_loc)
Processed search databases
KEGG db: None
KOfam db: None
KOfam KO list: None
UniRef db: None
Pfam db: None
dbCAN db: None
RefSeq Viral db: None
MEROPS peptidase db: None
VOGDB db: NoneDescriptions of search database entries
Pfam hmm dat: None
dbCAN family activities: None
VOG annotations: NoneDescription db: NoneDRAM distillation sheets
Genome summary form: None
Module step form: None
ETC module database: None
Function heatmap form: None
AMG database: None
喜欢开发的建议看一下这个文件:/envs/DRAM/lib/python3.10/site-packages/mag_annotator/database_handler.py
万能的帮助信息:
###help查看DRAM配置命令
DRAM-setup.py --helpusage: DRAM-setup.py [-h]{version,prepare_databases,set_database_locations,mv_db_folder,update_description_db,update_dram_forms,print_config,print_settings,import_config,export_config}...positional arguments:{version,prepare_databases,set_database_locations,mv_db_folder,update_description_db,update_dram_forms,print_config,print_settings,import_config,export_config}version print DRAM versionprepare_databases Download and process databases for annotationset_database_locationsSet database locations for already processed databasesmv_db_folder If you move a databases folder this will update all locations for all databases movedupdate_description_dbUpdate description databaseupdate_dram_forms Update DRAM distillate and liquor formsprint_config Print database locationsprint_settings Print database settingsimport_config Import CONFIG fileexport_config Export CONFIG fileoptions:-h, --help show this help message and exit
全新配置数据库:
###DRAM数据库设置,官网上写的是自己有kegg权限时的命令,估计大家更合适第二种
DRAM-setup.py prepare_databases --output_dir DRAM_data --kegg_loc kegg.pep#估计大家都没有kegg权限吧,github上写的是没有kegg权限时使用这个命令,
#这个如果使用已经有的数据库目录则需要再DRAM_data所在目录运行。
DRAM-setup.py prepare_databases --output_dir DRAM_data#或者直接将这里的DRAM_data写成已有DRAM_data目录所在的绝对路径,当然最终的DRAM_data目录名也可以根据自己实际修改:
DRAM-setup.py prepare_databases --output_dir /mnt/dramdata/DRAM_data########注意,这里不管使用有权限还是无权限的命令,都会在当前运行目录下生成一个DRAM_data的目录
###该DRAM_data目录下会有database_files文件夹和database_processing.log, 看字面意思就知道了吧
###一个是放kofam之类数据库的文件夹,一个是数据库处理的日志信息###查看数据处理日志database_processing.log
cat database_processing.log
2023-11-22 17:04:00,338 - Starting the process of downloading data
2023-11-22 17:04:00,339 - The gene_ko_link_loc argument was not used to specify a downloaded gene_ko_link file, and dram can not download it its self. So it is assumed that the user wants to set up DRAM without it
2023-11-22 17:04:00,339 - Database preparation started
2023-11-22 17:04:00,339 - Downloading kofam_hmm
2023-11-22 17:08:16,043 - Downloading kofam_ko_list
2023-11-22 17:08:18,051 - Downloading uniref##########################################
#这个uniref有41G,而且下载速度一般只有几百kb,所以大家得耐心等一下,
#或者提前下载这个文件到自己想要存放的文件夹下,同样可以先下载kofam_hmm和kofam_ko_list这两个文件,
#不过这两个文件下载速度都还可以,直接使用数据库设置命令就行了。
wget -c ftp://ftp.uniprot.org/pub/databases/uniprot/uniref/uniref90/uniref90.fasta.gz#但要注意的是运行数据库设置命令要在DRAM_data这个目录所在的目录下运行,如果已经进入到DRAM_data目录,
#则要 cd .. 回到上级目录,否则会自动在这个DRAM_data目录下再生成一个DRAM_data目录,原先下载的文件就看不到了。
#当然要避免这个问题需要想前面设置数据库命令中使用绝对路径。
DRAM-setup.py prepare_databases --output_dir /mnt/dramdata/DRAM_data导入导出原有数据库配置
###激活已有环境导出数据库配置
DRAM-setup.py export_config > my_config.json###在激活的新环境下导入数据库配置
DRAM-setup.py import_config --config_loc my_config.json官方的提示:NOTE: Setting up DRAM can take a long time (up to 5 hours) and uses a large about of memory (512 gb) by default. To use less memory you can use the --skip_uniref flag which will reduce memory usage to ~64 gb if you do not provide KEGG Genes and 128 gb if you do. Depending on the number of processors which you tell it to use (using the --threads argument) and the speed of your internet connection. On a less than 5 year old server with 10 processors it takes about 2 hours to process the data when databases do not need to be downloaded.
看到没? uniref消耗内存惊人!即使跳过uniref耗内存也要64gb和128gb,而且时间超级长,即使不需要下载数据库文件,也要10线程运行2个小时左右,有胖服务器的人来干吧,后面我再提供一下做好的数据库链接供大家下载。
直接设置已有数据库路径:
#设置示例:###以下是设置数据库路径的全部参数:
usage: DRAM-setup.py set_database_locations [-h] [--kegg_db_loc KEGG_DB_LOC][--kofam_hmm_loc KOFAM_HMM_LOC][--kofam_ko_list_loc KOFAM_KO_LIST_LOC][--uniref_db_loc UNIREF_DB_LOC][--pfam_db_loc PFAM_DB_LOC][--pfam_hmm_dat PFAM_HMM_DAT][--dbcan_db_loc DBCAN_DB_LOC][--dbcan_fam_activities DBCAN_FAM_ACTIVITIES][--vogdb_db_loc VOGDB_DB_LOC][--vog_annotations VOG_ANNOTATIONS][--viral_db_loc VIRAL_DB_LOC][--peptidase_db_loc PEPTIDASE_DB_LOC][--description_db_loc DESCRIPTION_DB_LOC][--genome_summary_form_loc GENOME_SUMMARY_FORM_LOC][--module_step_form_loc MODULE_STEP_FORM_LOC][--etc_module_database_loc ETC_MODULE_DATABASE_LOC][--function_heatmap_form_loc FUNCTION_HEATMAP_FORM_LOC][--amg_database_loc AMG_DATABASE_LOC][--update_description_db]optional arguments:-h, --help show this help message and exit--kegg_db_loc KEGG_DB_LOCmmseqs2 database file from kegg .pep file (default:None)--kofam_hmm_loc KOFAM_HMM_LOChmm file for KOfam, already processed with hmmpress(default: None)--kofam_ko_list_loc KOFAM_KO_LIST_LOCKOfam ko list file (default: None)--uniref_db_loc UNIREF_DB_LOCmmseqs2 database file from uniref .faa (default: None)--pfam_db_loc PFAM_DB_LOCmmseqs2 database file from pfam .hmm (default: None)--pfam_hmm_dat PFAM_HMM_DATpfam hmm .dat file to get PF descriptions (default:None)--dbcan_db_loc DBCAN_DB_LOChmm file for dbcan, already processed with hmmpress(default: None)--dbcan_fam_activities DBCAN_FAM_ACTIVITIESCAZY family activities file (default: None)--vogdb_db_loc VOGDB_DB_LOChmm file for vogdb, already processed with hmmpress(default: None)--vog_annotations VOG_ANNOTATIONSvog annotations file (default: None)--viral_db_loc VIRAL_DB_LOCmmseqs2 database file from ref seq viral genecollection (default: None)--peptidase_db_loc PEPTIDASE_DB_LOCmmseqs2 database file from MEROPS database (default:None)--description_db_loc DESCRIPTION_DB_LOCLocation to write description sqlite db (default:None)--genome_summary_form_loc GENOME_SUMMARY_FORM_LOCFile path to genome summary form (default: None)--module_step_form_loc MODULE_STEP_FORM_LOCFile path to module step form (default: None)--etc_module_database_loc ETC_MODULE_DATABASE_LOCFile path to etc module database (default: None)--function_heatmap_form_loc FUNCTION_HEATMAP_FORM_LOCFile path to function heatmap form (default: None)--amg_database_loc AMG_DATABASE_LOCFile path to amg database (default: None)--update_description_db额外数据库配置
除了kegg和uniref数据库外还可以使用额外数据库如甲基化数据库,不过官方说了需要1.4已上版本来使用这个功能:
#下载数据库文件
wget https://raw.githubusercontent.com/WrightonLabCSU/DRAM/master/data/methylotrophy/methylotrophy.faa
#将数据库注释
DRAM.py annotate -i '/some/path/*.fasta' -o dram_output --threads 30 --custom_db_name methyl --custom_fasta_loc methylotrophy.faa#To Distill with methyl
wget https://raw.githubusercontent.com/WrightonLabCSU/DRAM/master/data/methylotrophy/methylotrophy_distillate.tsv
DRAM.py distill -i dram_output/annotations.tsv -o dram_output/distillate --custom_distillate methylotrophy_distillate.tsv
3、运行DRAM
Once DRAM is set up you are ready to annotate some MAGs. The following commands will generate the full annotation and distillation of genomes:
基因组完整注释和提取
####DRAM注释
DRAM.py annotate -i 'my_bins/*.fa' -o annotation
DRAM.py distill -i annotation/annotations.tsv -o distill --trna_path annotation/trnas.tsv --rrna_path annotation/rrnas.tsv###以下是官方的说明:
my_bins should be replaced with the path to a directory which contains all of your bins you would like to annotate. If you only need to annotate a single genome (or an entire assembly) a direct path to a nucleotide fasta should be provided. Using 20 processors, DRAM.py takes about 17 hours to annotate ~80 MAGs of medium quality or higher from a mouse gut metagenome.###看到没?肠道宏基因组80个基因组需要20线程17个小时。##再附上输入输出说明:
DRAM annotate key parameters
Input files: Key input quality files
Input fasta: fasta file or a string with wildcards (e.g. ‘MAGs/*.fa’) that leads to multiple fastas
Bin taxonomy: a tab separated table with the first column being the names of the bins used as input to DRAM and another column with the header 'classification'. The output of GTDB-tk is already in this format and can be used directly.
Bin quality: a tab separated table with the first column being the names of the bins used as input to DRAM and additional columns with the headers 'Completeness' giving genome completeness information and 'Contamination' giving genome contamination information. The output of checkM is already in this format and can be used directly.
Output directory: Folder to be created that will store output files
Technical Parameters: These parameters effect results and resource use
Minimum contig size: Used for gene prediction, the defalt is 2500,
Minimum bit score for MMSeqs2 searches: 60
Minimum bit score for reverse best hit MMSeqs2 searches: 350
Number of threads to use: 10
Speed and Resources: These arguments make DRAM run faster but at a cost.
Use Uniref: Drastically decreases run time and memory requirements if set to False: True
Low Mem Mode, Skip annotating with uniref, use kofam over KEGG genes: False
Skip Trnascan: False
Troubleshooting: These additional arguments are popular for more specialized analyses.
Keep Tmp Dir', action='store_true', default=False)
Threads, number of processors to use: 10
Specialization: These additional arguments are popular for more specialized analyses.
Custom fasta Databases Locations: Used in blast style searches, see "Additional Databases" section.
Custom fasta Databases Names: Used in blast style searches, see "Additional Databases" section.
Custom HMM Databases Names: Used in HMM searches, see "Additional Databases" section.
Custom HMM Databases Locations: Used in HMM searches, see "Additional Databases" section.
Custom HMM Cutoff Locations: Used in HMM searches, see "Additional Databases" section.
GTDB taxonomy,'Summary file from gtdbtk taxonomy assignment from bins, can be used multiple times'
Less Popular: These arguments are rarely used, but you may need them in some cases
Mode of prodigal:
Translation table for prodigal.
DRAM annotate Outputs
Tab separated file (.tsv) with all the annotations from Pfam, KEGG, UniProt, dbCAN, and MEROPS databases for all genes in all the input genomes
GenBank files for each genome
Single gene-finding format (.gff) file of all annotations across genomes
Single fasta format file (.fasta) of each open reading frame nucleotide sequence and best ranked annotation (see Annotation grades section)
Single fasta format file (.fasta) of each translated open reading frame amino acid sequence and best ranked annotation KEGG annotation
Tab separated files (.tsv) with tRNAs and rRNAs使用示例:
Example 1.1
Suppose you have 2 FASTAs fasta_1.fa and fasta_2.fa you want to name them A_seqs and B_seqs respectively. You also have 2 hmms profils_cutoffs.hmm and profiles_bitscor.hmm, the first hmm you want to use custom cutoffs in a tsv file named cutoffs.tsv and the second you want to use bit score, you want to name them hmm_A, and hmm_B. The command that you would need to use is then.
# Assume all files are in the working directory.
DRAM.py annotate \-i <input path or regex> \-o <output path> \--threads 20 \--custom_fasta_loc ./fasta_1.fa \--custom_db_name A_seqs \--custom_fasta_loc ./fasta_2.fa \--custom_db_name B_seqs \--custom_hmm_name hmm_A \--custom_hmm_loc ./profils_cutoffs.hmm\--custom_hmm_cutoffs_loc ./cutoffs.tsv \--custom_hmm_name hmm_B \--custom_hmm_loc ./profiles_bitscor.hmm # this hmm will use bitscores
Again, the order of the arguments determines the interpretation.
DRAM Annotate Called Genes
You can annotate already called genes in DRAM, if necessary, and get most of the same output, the gff output will not be generated for example but annotations will be made and distillation will be possible. You may want to check that the names of your genes are to your liking in the FAA file as those names will be carried over and may be changed by the software that you generated the file with, including Dram. You should also note that this command works on only one faa at a time. As of the 1.3 release custom HMMs are not supported though custom FASTAs are. The commands are otherwise the same. See the example below.
Example 1.2
Suppose you have a FASTA of Amino Acid Sequences fasta.faa. The command that you would need to use is then.
# Assume all files are in the working directory.
DRAM.py annotate-genes \-i <input path to the genes faa, NO REGEX> \-o <output path> \--threads 20 \--custom_fasta_loc ./fasta_1.fa \--custom_db_name A_seqs \--custom_fasta_loc ./fasta_2.fa \--custom_db_name B_seqs \
DRAM distill
After your annotation is finished, you can summarize these annotations with the following command:
DRAM.py distill -i annotation/annotations.tsv -o genome_summaries --trna_path annotation/trnas.tsv --rrna_path annotation/rrnas.tsvThis command will generate three files. The first is called genome_summary.xlsx which contains a summary of metabolisms present in each genome. It gives gene by gene information across various metabolisms for every genome in your dataset. The genome_statistics.tsv file contains all measures required by the MIMAG about each fasta used as input. Finally, the liquor.html is an interactive html that allows users to hover over each box to see what genes prompted the box color (Example here) and was manually curated to consider alternate genes for pathways and single processes. This heat map allows the user to quickly profile ecosystem relevant processes across hundreds of genomes.
DRAM strainer
After you have completed your annotation and distillation, you may want to further analyze genes of interest by making trees or functional modeling. To pull the genes you can use DRAM.py strainer. For example, if you want to pull all pmoa/amoa genes based on KEGG annotations you can make a tree:
DRAM.py strainer --identifiers K10944 -i annotations.tsv -f genes.faa -o amoa_pmoa_genes.faaOr you might want to blast a few specific genes:
DRAM.py strainer --genes bin.2_scaffold_2_3 bin.4_scaffold_12_42 -i annotations.tsv -f genes.dna -on my_genes.fnaOr maybe you only want to see genes that are involved in glycolysis or the TCA cycle that are from bins from the Roseburia genus:
DRAM.py strainer -i hmp_bins/annotations.tsv -f hmp_bins/genes.fna -o genes.roseburia.glycoloysis_tca.fna --taxonomy g__Roseburia --categories glycolysis TCADRAM strainer parameters
- Input files:
- Annotations: annotations.tsv file generated during the annotate step
- Input fasta: genes fasta file (.faa or .fna) to be filtered
- Output fasta: location to save filtered fasta file
- Default Parameters:
- Fastas: None
- Scaffolds: None
- Genes: None
- Identifiers: None
4、运行DRAM-v
The contigs must be processed with VirSorter and the processed viral contigs and VIRSorter_affi-contigs.tab are used as input to DRAM-v. The following commands will generate the full annotation and distillation of viral contigs:
基于virsorter的结果进行注释,相当于过滤了。
所以其实这里还需要安装virsorter软件,virsorter的操作使用后面再补上
DRAM-v.py annotate -i my_viral_contigs.fa -v VIRSorter_affi-contigs.tab -o annotation##In the output annotation folder there will be the same collection of files as is generated when running DRAM.py. The only change is the addition of columns for VIRSorter gene category, auxiliary score and metabolic flags to the annotations.tsv output file.DRAM-v.py distill -i annotation/annotations.tsv -o annotation/distilledDRAM-v strainer
DRAM-v strainer filters down fasta files to only contain genes or scaffolds that meet criteria set by the user. For example, if you want to get the amino acid sequences of all genes annotated with the GH4 and GH5 families in fasta format to make a tree then you can do that with this command:
DRAM-v.py strainer --identifiers GH4 GH5 -i annotations.tsv -f genes.faa -o GH4_GH5_genes.faaOr if you want to only get scaffolds 34 and 52 from the fasta bin.4 then you can use this command:
DRAM-v.py strainer --scaffolds bin.4_scaffold_34 bin.4_scaffold_52 -i annotations.tsv -f scaffolds.fna -o GH4_GH5_genes.faaIf you only want to get the genes considered potential AMGs that are transporters use this command:
DRAM-v.py strainer -i hmp_viruses/annotations.tsv -f hmp_viruses/genes.fna -o genes.amgs.fna -a --categories TransportersIf I only want genes that have a 'V' flag and auxiliary scores of 4 or 5:
DRAM-v.py strainer -i hmp_viruses/annotations.tsv -f hmp_viruses/genes.fna -o genes.viral.fna --aux_scores 4 5 --amg_flags VDRAM-v strainer parameters
- Input files
- Annotations: annotations.tsv file generated during the annotate step
- Input fasta: genes fasta file (.faa or .fna) to be filtered
- Output fasta: location to save filtered fasta file
- Default Parameters
- fastas: None
- Scaffolds: None
- Genes: None
- Identifiers: None
5、系统资源需求:
DRAM has a large memory burden and is designed to be run on high performance computers. DRAM annotates against a large variety of databases which must be processed and stored. Setting up DRAM with KEGG Genes and UniRef90 will take up ~500 GB of storage after processing and require ~512 GB of RAM while using KOfam and skipping UniRef90 will mean all processed databases will take up ~30 GB on disk and will only use ~128 GB of RAM while processing. DRAM annotation memory usage depends on the databases used. When annotating with UniRef90 around 220 GB of RAM is required. If the KEGG gene database has been provided and UniRef90 is not used then memory usage is around 100 GB of RAM. If KOfam is used to annotate KEGG and UniRef90 is not used then less than 50 GB of RAM is required. DRAM can be run with any number of processors on a single node.
DRAM annotate functions are only tested in linux and DRAM distillation functions are tested in linux and macOS environments. It will likely work in OSX environments. It is unlikely to work in Windows.
看不懂的百度翻译吧,反正需求内存不小。
DRAM具有很大的内存负担,并且被设计为在高性能计算机上运行。DRAM针对必须处理和存储的各种数据库进行注释。使用KEGG Genes和UniRef90设置DRAM在处理后将占用约500 GB的存储空间,并且在使用KOfam时需要约512 GB的RAM。跳过UniRef90将意味着所有处理后的数据库将占用约30 GB的磁盘空间,在处理时仅使用约128 GB的RAM。DRAM注释内存的使用情况取决于所使用的数据库。使用UniRef90进行注释时,需要大约220 GB的RAM。如果已经提供了KEGG基因数据库,并且没有使用UniRef90,那么存储器的使用量大约是100GB的RAM。如果使用KOfam对KEGG进行注释,而未使用UniRef90,则需要少于50 GB的RAM。DRAM可以在单个节点上与任何数量的处理器一起运行。
DRAM注释函数仅在linux中测试,DRAM提取函数在linux和macOS环境中测试。它可能在OSX环境中工作。它不太可能在Windows中工作。
这篇关于DRAM(Distilling and Refining Annotations of Metabolism,提取和精练代谢注释)工具安装和使用的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!




