本文主要是介绍ChAMP分析TCGA结直肠癌甲基化数据实战,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
前面用几篇推文详细介绍了ChAMP包用于甲基化分析的流程,并使用肠癌领域的GSE149282进行了演示。
16.ChAMP分析甲基化数据:标准流程
17.ChAMP分析甲基化数据:从β值矩阵开始的流程
下面我们用TCGA-COAD和TCGA-READ的甲基化数据再做一次演示,从IDAT文件开始。
有了这个结果之后,你就可以去做各种联合分析~
太长不看版
下面的步骤都是在服务器上做的,因为一共有812个样本,至少需要64+G的内存,一般的个人电脑就不要尝试了,需要这个分析结果的可以私聊我,有偿获取!
去掉日志文件就是做了以下几个分析:
library(ChAMP)myDir="./coreadidatAllinone"
myLoad <- champ.load(myDir)
save(myLoad,file="coread_methy_myload.rdata")myNorm <- champ.norm(beta = myLoad$beta,cores = 20)
save(myNorm,file = "coread_methy_mynorm.rdata")champ.SVD(beta = myNorm |> as.data.frame(), pd=myLoad$pd)myDMP <- champ.DMP()
myDMR <- champ.DMR()
save(myDMP,myDMR,file = "coread_methy_dmpr.rdata")myGSEA <- champ.GSEA()
save(myGSEA,file = "coread_methy_gsea.rdata")
下面是分步版本,日志文件太多了…
数据准备
数据下载可以用之前介绍过的TCGAbiolinks包,也可以直接去GDC官网下载。
下载后把所有的IDAT文件放在一个文件夹中:
我是用下面的代码,用什么方法都行,只要把所有的IDAT放在一个文件夹下就可以了。
lapply(list.files("./coread_idat/",recursive = T,pattern = "idat$",full.names = T),file.copy, to = "./coreadidatAllinone/")
然后在GDC官网下载gdc_sample_sheet这个文件,这个文件可以帮助我们制作自己的样本信息csv文件。
sample_sheet <- read.table("gdc_sample_sheet.2022-08-21.tsv",sep = "\t",header = T)
之后按照CnAMP包的要求制作csv文件,可以参考历史推文:xxxxxxxxxxx
pd <- sample_sheet[,c("File.Name","Project.ID","Sample.ID","Sample.Type")]
pd$Sentrix_ID <- substr(pd$File.Name,1,36)
pd$Sentrix_Position <- substr(pd$File.Name,38,41)
pd$Sample_Group <- ifelse(pd$Sample.Type == "Solid Tissue Normal","normal","tumor")
pd <- pd[,c(3,7,2,5,6)]
names(pd)[c(1,3)] <- c("Sample_Name","Project")pd1 <- pd[duplicated(pd$Sentrix_ID),]
把这个文件保存到coreadidatAllinone这个文件夹下即可:
write.csv(pd1,file = "./coreadidatAllinone/sample_sheet.csv",quote = F,row.names = F)
读取数据
# 加载R包
#suppressMessages(library(ChAMP))
library(ChAMP)
非常简单,指定合适的路径,里面有IDAT文件和相应的样本信息csv文件,就不会出错。
# 指定文件夹路径
myDir="./coreadidatAllinone"myLoad <- champ.load(myDir)
下面是加载日志文件:
[===========================]
[<<<< ChAMP.LOAD START >>>>>]
-----------------------------[ Loading Data with ChAMP Method ]
----------------------------------
Note that ChAMP method will NOT return rgSet or mset, they object defined by minfi. Which means, if you use ChAMP method to load data, you can not use SWAN or FunctionNormliazation method in champ.norm() (you can use BMIQ or PBC still). But All other function should not be influenced.[===========================]
[<<<< ChAMP.IMPORT START >>>>>]
-----------------------------[ Section 1: Read PD Files Start ]CSV Directory: ./coreadidatAllinone/sample_sheet.csvFind CSV SuccessReading CSV FileReplace Sentrix_Position into ArrayReplace Sentrix_ID into SlideThere is NO Pool_ID in your pd file.There is NO Sample_Plate in your pd file.There is NO Sample_Well in your pd file.
[ Section 1: Read PD file Done ][ Section 2: Read IDAT files Start ]Loading:./coreadidatAllinone/4edebe4a-9fb7-4d84-a260-97feb38fb16a_noid_Grn.idat ---- (1/406)
Warning in readChar(con, nchars = n) :truncating string with embedded nulsLoading:./coreadidatAllinone/25df3fb5-44fe-41b0-92ad-cf30bdb62584_noid_Grn.idat ---- (2/406)
Warning in readChar(con, nchars = n) :truncating string with embedded nulsLoading:./coreadidatAllinone/f946a265-a27e-4c20-bd53-266ab6aa3de6_noid_Grn.idat ---- (3/406)Loading:./coreadidatAllinone/4220d653-e946-4741-8864-cc8246939003_noid_Red.idat ---- (405/406)
Warning in readChar(con, nchars = n) :truncating string with embedded nulsLoading:./coreadidatAllinone/b9623ff8-8fd4-4124-bd08-b128767ea60a_noid_Red.idat ---- (406/406)
Warning in readChar(con, nchars = n) :truncating string with embedded nuls
## ........Extract Mean value for Green and Red Channel SuccessYour Red Green Channel contains 622399 probes.
[ Section 2: Read IDAT Files Done ][ Section 3: Use Annotation Start ]Reading 450K Annotation >>Fetching NEGATIVE ControlProbe.Totally, there are 613 control probes in Annotation.Your data set contains 613 control probes.Generating Meth and UnMeth MatrixExtracting Meth Matrix...Totally there are 485512 Meth probes in 450K Annotation.Your data set contains 485512 Meth probes.Extracting UnMeth Matrix...Totally there are 485512 UnMeth probes in 450K Annotation.Your data set contains 485512 UnMeth probes.Generating beta MatrixGenerating M MatrixGenerating intensity MatrixCalculating Detect P valueCounting Beads
[ Section 3: Use Annotation Done ][<<<<< ChAMP.IMPORT END >>>>>>]
[===========================]
[You may want to process champ.filter() next.][===========================]
[<<<< ChAMP.FILTER START >>>>>]
-----------------------------In New version ChAMP, champ.filter() function has been set to do filtering on the result of champ.import(). You can use champ.import() + champ.filter() to do Data Loading, or set "method" parameter in champ.load() as "ChAMP" to get the same effect.This function is provided for user need to do filtering on some beta (or M) matrix, which contained most filtering system in champ.load except beadcount. User need to input beta matrix, pd file themselves. If you want to do filterintg on detP matrix and Bead Count, you also need to input a detected P matrix and Bead Count information.Note that if you want to filter more data matrix, say beta, M, intensity... please make sure they have exactly the same rownames and colnames.[ Section 1: Check Input Start ]You have inputed beta,intensity for Analysis.pd file provided, checking if it's in accord with Data Matrix...pd file check success.Parameter filterDetP is TRUE, checking if detP in accord with Data Matrix...detP check success.Parameter filterBeads is TRUE, checking if beadcount in accord with Data Matrix...beadcount check success.parameter autoimpute is TRUE. Checking if the conditions are fulfilled...!!! ProbeCutoff is 0, which means you have no needs to do imputation. autoimpute has been reset FALSE.Checking Finished :filterDetP,filterBeads,filterMultiHit,filterSNPs,filterNoCG,filterXY would be done on beta,intensity.You also provided :detP,beadcount .
[ Section 1: Check Input Done ][ Section 2: Filtering Start >>Filtering Detect P value StartThe fraction of failed positions per sampleYou may need to delete samples with high proportion of failed probes:Failed CpG Fraction.
TCGA-DC-6158-01A 0.0036394569
TCGA-F5-6864-01A 0.0170891760
TCGA-EI-6917-01A 0.0266790522
TCGA-AF-2690-01A 0.0019216827
TCGA-EI-6513-01A 0.0011801974
TCGA-EI-6885-01A 0.0056970785
TCGA-DT-5265-01A 0.0030545074
TCGA-AG-A036-11A 0.0004613686
TCGA-F5-6810-01A 0.0058433159
TCGA-EI-6506-01A 0.0021276508
TCGA-EI-6510-01A 0.0025993178
TCGA-EI-7002-01A 0.0132252138
## ...
TCGA-DM-A28C-01A 0.0013367332
TCGA-A6-5664-01A 0.0021297105
TCGA-D5-6533-01A 0.0019093246Filtering probes with a detection p-value above 0.01.Removing 59892 probes.If a large number of probes have been removed, ChAMP suggests you to identify potentially bad samplesFiltering BeadCount StartFiltering probes with a beadcount <3 in at least 5% of samples.Removing 205 probesFiltering NoCG StartOnly Keep CpGs, removing 1636 probes from the analysis.Filtering SNPs StartUsing general 450K SNP list for filtering.Filtering probes with SNPs as identified in Zhou's Nucleic Acids Research Paper 2016.Removing 47713 probes from the analysis.Filtering MultiHit StartFiltering probes that align to multiple locations as identified in Nordlund et alRemoving 10 probes from the analysis.Filtering XY StartFiltering probes located on X,Y chromosome, removing 8124 probes from the analysis.Updating PD fileFixing Outliers StartReplacing all value smaller/equal to 0 with smallest positive value.Replacing all value greater/equal to 1 with largest value below 1..
[ Section 2: Filtering Done ]All filterings are Done, now you have 367932 probes and 406 samples.[<<<<< ChAMP.FILTER END >>>>>>]
[===========================]
[You may want to process champ.QC() next.][<<<<< ChAMP.LOAD END >>>>>>]
[===========================]
[You may want to process champ.QC() next.]
save(myLoad,file="coread_methy_myload.rdata")
预处理
# 数据预处理,聚类树由于样本太多显示不出来会报错
champ.QC()
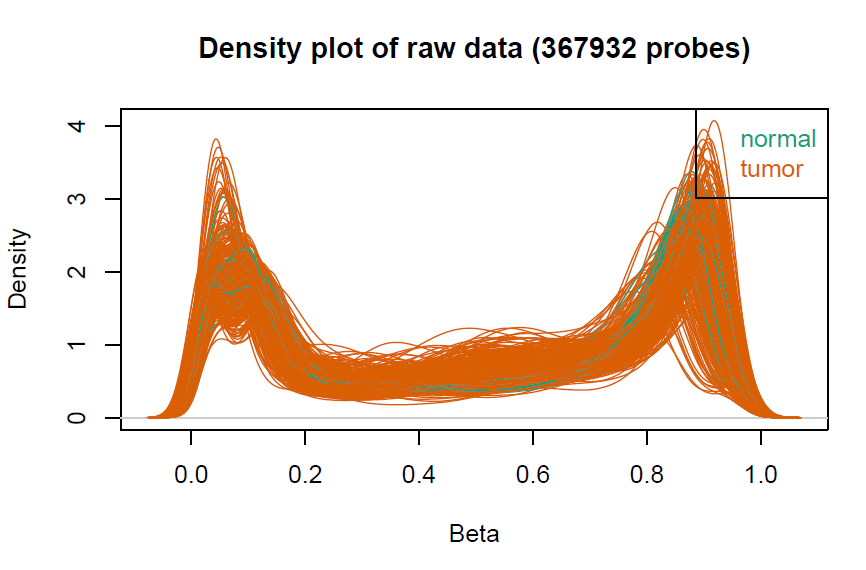
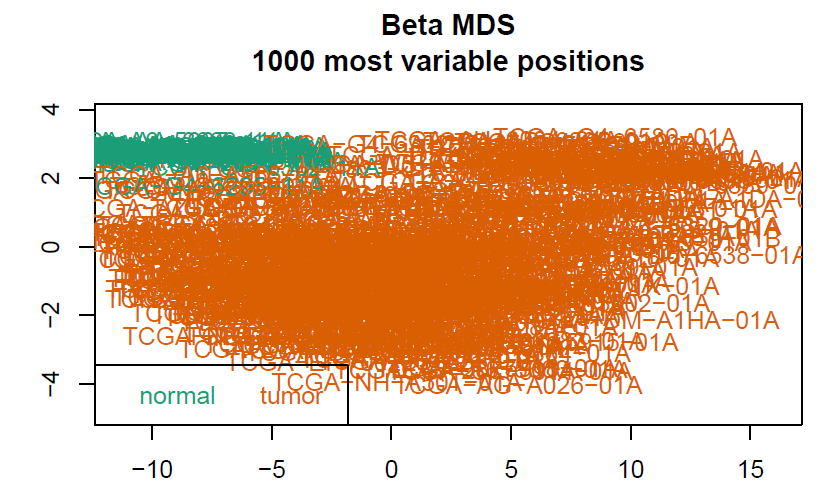
myNorm <- champ.norm(beta = myLoad$beta,cores = 20)
日志文件:
[===========================]
[>>>>> ChAMP.NORM START <<<<<<]
-----------------------------
champ.norm Results will be saved in ./CHAMP_Normalization/
[ SWAN method call for BOTH rgSet and mset input, FunctionalNormalization call for rgset only , while PBC and BMIQ only needs beta value. Please set parameter correctly. ]Note that,BMIQ function may fail for bad quality samples (Samples did not even show beta distribution).
20 cores will be used to do parallel BMIQ computing.
[>>>>> ChAMP.NORM END <<<<<<]
[===========================]
[You may want to process champ.SVD() next.]
save(myNorm,file = "coread_methy_mynorm.rdata")
champ.SVD(beta = myNorm |> as.data.frame(), # 这里需要注意pd=myLoad$pd)
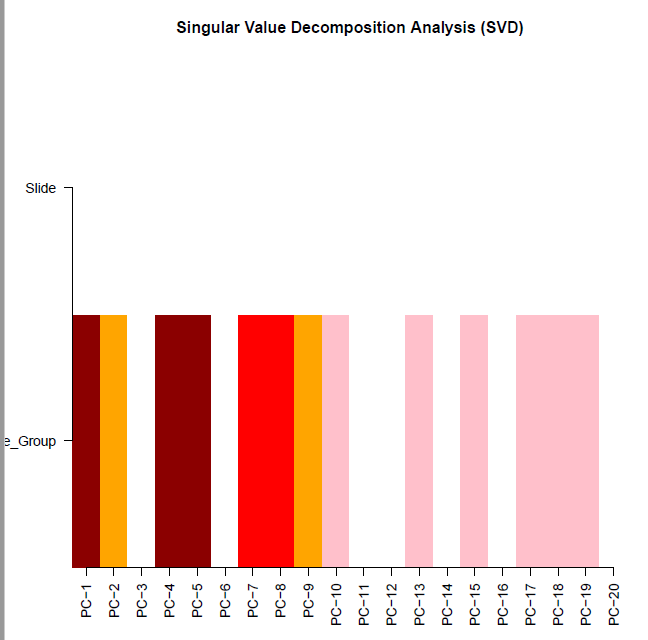
[===========================]
[<<<<< ChAMP.SVD START >>>>>]
-----------------------------
champ.SVD Results will be saved in ./CHAMP_SVDimages/ .Your beta parameter is data.frame format. ChAMP is now changing it to matrix.
[SVD analysis will be proceed with 367932 probes and 406 samples.][ champ.SVD() will only check the dimensions between data and pd, instead if checking if Sample_Names are correctly matched (because some user may have no Sample_Names in their pd file),thus please make sure your pd file is in accord with your data sets (beta) and (rgSet).]<Sample_Group>(character):tumor, normal
<Slide>(character):4edebe4a-9fb7-4d84-a260-97feb38fb16a, 25df3fb5-44fe-41b0-92ad-cf30bdb62584, f946a265-a27e-4c20-bd53-266ab6aa3de6, 50348bd8-7c8d-47d8-9d95-a1e20e5decc8, f3bd946a-f42f-4343-8de5-6092ab6de36b, 57628681-9510-4bdc-8183-c3ae5fbf5b6b, 95b971ce-8e6f-46dc-ab85-a7b729cc9944, 1ffe442d-6bc6-445e-8b68-0d3ed6552dec, 98a4f10a-a045-492a-a621-fe1f3b117103, 5d6285d0-d714-4c2e-836f-47dc65ad598c, 1679e880-0db3-4a64-9a40-9f962314aa9e,
##.....
2853c315-b9f3-409e-bb60-25b05a5cfa64, 39cb9c2e-63a8-4513-a5f8-a657f4ec43b6, d16d1d2e-6ebf-4b72-8840-fcc9d3c2dd52, 4220d653-e946-4741-8864-cc8246939003, b9623ff8-8fd4-4124-bd08-b128767ea60a
[champ.SVD have automatically select ALL factors contain at least two different values from your pd(sample_sheet.csv), if you don't want to analysis some of them, please remove them manually from your pd variable then retry champ.SVD().]<Sample_Name>
<Project>
<Array>
[Factors are ignored because they only indicate Name or Project, or they contain ONLY ONE value across all Samples.][<<<<<< ChAMP.SVD END >>>>>>]
[===========================]
[If the batch effect is not significant, you may want to process champ.DMP() or champ.DMR() or champ.BlockFinder() next, otherwise, you may want to run champ.runCombat() to eliminat batch effect, then rerun champ.SVD() to check corrected result.]Sample_Group Slide[1,] 2.289393e-26 0.4906548[2,] 3.959362e-03 0.4906548[3,] 8.836284e-01 0.4906548[4,] 7.938020e-17 0.4906548[5,] 3.373293e-14 0.4906548[6,] 9.808115e-01 0.4906548[7,] 4.995617e-08 0.4906548[8,] 1.220760e-06 0.4906548[9,] 1.619588e-04 0.4906548
[10,] 2.288907e-02 0.4906548
[11,] 6.407854e-01 0.4906548
[12,] 5.390941e-02 0.4906548
[13,] 1.308536e-02 0.4906548
[14,] 6.585889e-01 0.4906548
[15,] 2.557036e-02 0.4906548
[16,] 5.390941e-02 0.4906548
[17,] 2.045547e-02 0.4906548
[18,] 4.091545e-02 0.4906548
[19,] 1.601403e-02 0.4906548
[20,] 6.241584e-01 0.4906548
差异分析
myDMP <- champ.DMP()
日志文件:
[===========================]
[<<<<< ChAMP.DMP START >>>>>]----------------------------
!!! Important !!! New Modification has been made on champ.DMP(): (1): In this version champ.DMP() if your pheno parameter contains more than two groups of phenotypes, champ.DMP() would do pairewise differential methylated analysis between each pair of them. But you can also specify compare.group to only do comparasion between any two of them.(2): champ.DMP() now support numeric as pheno, and will do linear regression on them. So covariates like age could be inputted in this function. You need to make sure your inputted "pheno" parameter is "numeric" type.--------------------------------[ Section 1: Check Input Pheno Start ]You pheno is character type.Your pheno information contains following groups. >><tumor>:363 samples.<normal>:43 samples.[The power of statistics analysis on groups contain very few samples may not strong.]pheno contains only 2 phenotypescompare.group parameter is NULL, two pheno types will be added into Compare List.tumor_to_normal compare group : tumor, normal[ Section 1: Check Input Pheno Done ][ Section 2: Find Differential Methylated CpGs Start ]-----------------------------Start to Compare : tumor, normalContrast MatrixContrasts
Levels ptumor-pnormalpnormal -1ptumor 1You have found 212017 significant MVPs with a BH adjusted P-value below 0.05.Calculate DMP for tumor and normal done.[ Section 2: Find Numeric Vector Related CpGs Done ][ Section 3: Match Annotation Start ][ Section 3: Match Annotation Done ][<<<<<< ChAMP.DMP END >>>>>>]
[===========================]
[You may want to process DMP.GUI() or champ.GSEA() next.]
myDMR <- champ.DMR()
日志文件:
[===========================]
[<<<<< ChAMP.DMR START >>>>>]
-----------------------------
!!! important !!! We just upgrate champ.DMR() function, since now champ.DMP() could works on multiple phenotypes, but ProbeLasso can only works on one DMP result, so if your pheno parameter contains more than 2 phenotypes, and you want to use ProbeLasso function, you MUST specify compare.group=c("A","B"). Bumphunter and DMRcate should not be influenced.[ Section 1: Check Input Pheno Start ]You pheno is character type.Your pheno information contains following groups. >><tumor>:363 samples.<normal>:43 samples.[ Section 1: Check Input Pheno Done ][ Section 2: Run DMR Algorithm Start ]Loading required package: IlluminaHumanMethylation450kanno.ilmn12.hg19
3 cores will be used to do parallel Bumphunter computing.
According to your data set, champ.DMR() detected 6693 clusters contains MORE THAN 7 probes within300 maxGap. These clusters will be used to find DMR.[bumphunterEngine] Parallelizing using 3 workers/cores (backend: doParallelMC, version: 1.0.17).
[bumphunterEngine] Computing coefficients.
[bumphunterEngine] Smoothing coefficients.
Loading required package: rngtools
[bumphunterEngine] Performing 250 bootstraps.
[bumphunterEngine] Computing marginal bootstrap p-values.
[bumphunterEngine] Smoothing bootstrap coefficients.
[bumphunterEngine] cutoff: 0.473
[bumphunterEngine] Finding regions.
[bumphunterEngine] Found 3289 bumps.
[bumphunterEngine] Computing regions for each bootstrap.
[bumphunterEngine] Estimating p-values and FWER.
Bumphunter detected 1226 DMRs with P value <= 0.05.[ Section 2: Run DMR Algorithm Done ][<<<<<< ChAMP.DMR END >>>>>>]
[===========================]
[You may want to process DMR.GUI() or champ.GSEA() next.]
save(myDMP,myDMR,file = "coread_methy_dmpr.rdata")
富集分析
# 富集分析
myGSEA <- champ.GSEA()
日志文件:
[===========================]
[<<<< ChAMP.GSEA START >>>>>]
-----------------------------
<< Prepare Gene List Ready >>
<< Start Do GSEA on each Gene List >>
<< Do GSEA on Gene list DMP>>
<< Pale Fisher Exact Test will be used to do GSEA >><< The category information is downloaded from MsigDB, and only simple Fisher Exact Test will be used to calculate GSEA. This method is suitable if your genes has equalivalent probability to be enriched. If you are using CpGs mapping genes, gometh method is recommended.>>
<< Done for Gene list DMP >>
<< Do GSEA on Gene list DMR>>
<< Pale Fisher Exact Test will be used to do GSEA >><< The category information is downloaded from MsigDB, and only simple Fisher Exact Test will be used to calculate GSEA. This method is suitable if your genes has equalivalent probability to be enriched. If you are using CpGs mapping genes, gometh method is recommended.>>
<< Done for Gene list DMR >>
[<<<<< ChAMP.GSEA END >>>>>>]
[===========================]
save(myGSEA,file = "coread_methy_gsea.rdata")
ChAMP实在是太简单了,不管多少样本都是几行代码完事,你值得拥有!
这篇关于ChAMP分析TCGA结直肠癌甲基化数据实战的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!






