本文主要是介绍Molecule Property Prediction based on Spatial Graph Embedding | 基于空间图嵌入的分子性质预测 | Graph | GNN,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
文章在2019.8发表在JCIM上,化学二区
1 Introduction
1.1 背景
目前深度学习方法 过度依赖手工设计的特征,这可能会限制模型 潜在表征 的搜索空间,那么如何设计卷积方法 来提高分子性质预测的性能 是需要解决问题。
1.2 本文工作
本文提出了一种 卷积空间图嵌入层(C-SGEL),使卷积运算能够应用于 分子图数据,将多个嵌入层堆叠起来 构造 卷积空间图嵌入网络(C-SGEN),用于从分子图中学习特征。此外,使用分子指纹来提高特征的泛化性能,并设计了一个结合 图形特征和指纹的复合模型 来预测分子性质。
2 模型介绍
2.1 模型结构
①在本文模型中,每个分子分别用无向图和分子指纹来表示。具体实现了a图表示层、b图嵌入层、c卷积空间图嵌入层 和 d图聚集层 四个过程。此外,结合这四个过程,提出了卷积空间图嵌入网络(C-SGEN),将CNN应用于分子图数据。此外,使用skip connection解决卷积过程中的梯度消失问题,在每个 卷积空间图嵌入层 C-SGEL 之后,引入跳过连接来连接每个C-SGEL的输出,它可以充分利用分子信息,揭示分子中原子之间的关系。
②同时,使用DNN处理的分子指纹。
③最后,将分子图和指纹表示连接在一起,使用全连接层预测分子特性。
模型结构图如下:’

2.2 分子图的表示
每个分子图G由 邻接矩阵A 和 初始节点矩阵X 表示。
①邻接矩阵A 表示原子之间的连接信息,若两个原子之间有边相连,则Aij =1,否则,Aij = 0,同时,邻接矩阵也考虑了原子的自连接。
②初始节点矩阵X 表示n个节点的集合,即 X = { x1, x2, … , xn },每个节点是一个m维向量,即 xi = { xi1, xi2, … , xim }。原子的特征描述如下:

xij 是第j个特征的值,m是特征维度
通过DeepChem软件包提取一个分子的所有特征,并编码到one-hot中。
(e.g)分子的初始特征矩阵。提取异丙醇的简单原子属性,不同颜色对应于不同原子。

每个原子由77维的向量构成,one-hot编码,这些向量构成了矩阵X。
选择顺序:矩阵中原子的顺序对模型的性能至关重要。选择原子的度作为原子顺序的标准。由于分子图属于无向图,因此分子中每个原子的度数是其相邻原子的数量。在本文中,分子中的初始特征矩阵按原子 度减少 的顺序排序。
不考虑氢原子,所以图中只有四个原子
2.3 图嵌入层
在得到分子的图表示之后,使用图嵌入层 得到 空间图形矩阵。
①由于one-hot表示比较稀疏,可能导致维度灾难,因此要首先对原子特征矩阵进行降维,X0 是初始矩阵,XF 是降维后得到的矩阵:
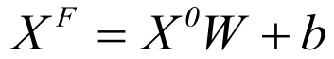
②随后,计算原子的空间图形矩阵 XS ,它包含原子与其相邻原子之间的连接信息,其中XFi 表示 XF 的第i行。XF 与 XS 均作为 C-SGEL的输入。XS 的计算如下:
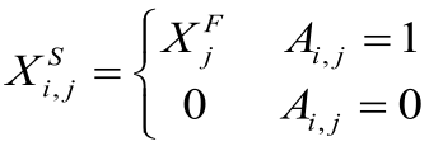
维度灾难:通常是指在涉及到向量的计算的问题中,随着维数的增加,计算量呈指数倍增长的一种现象。
①W是可学习的权重矩阵,b是可学习的偏差 XF : n × d1 X0:n × d0 W:d0 × d1 b:n × d1 d1
是转换后的维度,d0 是转换前的维度,d1<d0
②XS :n×n×d
实际上,XS 是考虑到与其他原子之间的空间连接的每个原子的特征。若两个原子之间有连接,那么这个位置就用原子的特征表示,否则就用0向量表示。
为了对分子图数据进行规则卷积运算,实现端到端的学习,需要确定分子的邻接矩阵和特征矩阵的大小。本文将邻接矩阵初始化为零,并将原子数设置为数据集中总原子数的平均值。
(e.g)以异丙醇为例,分子的初始 空间 图矩阵如下,其中A是邻接矩阵,XF 是降维后的分子特征矩阵,XS 是空间图矩阵。

X1S :代表第一层,这一层就代表了 自己 与 自己的邻居 的特征的矩阵 (一层 是 一个原子的 初始空间图矩阵)
XS :n×n×d,对应邻接矩阵A,若两个原子之间有连接,也就是A中为1的位置,那么在XS 中这个位置就用原子的特征表示(坐标是(i,j),用原子j的特征表示),否则就用0向量表示。(所有 原子的 初始空间图矩阵)
2.4 卷积空间图嵌入层(C-SGEL)
假设卷积核大小k=3,为了保持原子特征维度,第一个卷积层的kernel、padding、stride大小分别为3、1、1,本层结构如下:

C-SGEL 主要用来处理 空间图矩阵XSi 与 特征向量 XFi 。
① 首先,对于每个原子,对原子空间图矩阵XSi 卷积得到 Xik ,k是1D卷积的通道数,是一个超参数,它决定了后续1D卷积的核大小。
② 更新原子的特征向量:将原子i的特征向量xif 与Xik 拼起来,输入到两个1D卷积中,这样,每个原子表示的更新都考虑了 原子本身的信息 与 邻居的信息。
③ 拼接:更新后的原子特征向量与初始原子特征向量连接,这样就可以学习相邻原子的信息,并专注于自己本身的信息。
④ 堆叠多个 卷积空间图嵌入层(C-SGEL)以获得 卷积空间图嵌入网络(C-SGEN),如图1的椭圆部分。每个C-SGEL(每一层)可以从直接相邻原子中提取信息,因此C-SGEN(整个网络)可以提取原子的多阶邻域信息,通过重复C-SGEL计算得到更新的原子表示。与通道数k一样,C-SGEL的层数也是一个超参数。
XS 的第i层 与 XF 的第i行
2.5 图聚集层
为了增强原子与周围信息的集成能力,并且对特征排序的变化不敏感,本文使用所有原子的特征之和来表示一个图,如下:

2.6 DNN for fingerprint
为了保证分子特征的泛化性,分子指纹作为网络的另一个输入。本文使用ChemoPy30提取分子指纹,使用DNN处理获得的指纹。分子指纹表示为xfinger 。
ChemoPy30:是一个强大的Python软件包,用于计算 化学信息学 领域 的 大量 分子描述符。
DNN : 多层感知机,有很多隐藏层的神经网络
x finger :1×d
2.7 全连接层预测分子性质
将 卷积空间嵌入层得到的 xgraph 与 分子指纹得到的 xfinger_optimal 拼接在一起的到最终的分子表示 xmolecular ,即 xmolecular = [xgraph , xfinger_optimal ] 。最后通过全连接层预测分子性质,即

3 实验
3.1 数据集
比例:8:1:1
从DeepChem中获取公开可用的基准数据集,分别是ESOL、FreeSolv、Lipophilicity、PDBbind-full 、PDBbind-redefined
(为了与 MoleculeNet32 中的 benchmarks 进行比较,将 ESOL、FreeSolv 和 Lipophilicity 随机拆分,PDBbind 进行时间拆分。所有数据集分为三组:训练集(80%)、验证集(10%)和测试集(10%),然后在模型上固定不同的数值种子来评估性能。此外,对于不同的数据集,我们还通过实验确定了模型的超参数。)
Deepchem含有大量的内置数据集,主要是一些分子的数据,所以,数据集的名字为:MoleculeNet (分子网络)
ESOL:由实验水溶性数据组成。(由1128种水溶解度数据组成,直接从化合物的结构计算。化合物以SMILES表示,因此不包含分子中原子空间排列的信息)
FreeSolv:是水中水合自由能的数据集。(结构信息包含在SMILES字符串中)
Lipophilicity:亲脂性数据集,指的是分子在非极性溶剂中的溶解能力。
PDBbind-full:药物靶标亲和力数据集,包含约15000个数据。
PDBbind-redefined:在PDBlind的基础上提取4000条质量更好的数据。RMSE表示预测值与实验值之间的偏差,计算方法是MSE取根号 ,RMSE值越小,预测模型的性能越好。
3.2 评估指标
RMSE,计算方法为:
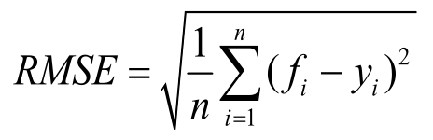
3.3 不同的指纹处理方式
分别采用DNN与CNN来处理分子指纹,同时增加了不经过任何处理的对照组,实验结果如下:

纵向观察,RMSE越小越好,故DNN效果最好,因此本文采用了DNN处理分子指纹
3.4 与其他模型的比较
ESOL、FreeSolv与Lipophilicity数据集:
在这三个数据集(物理化学任务)上,test阶段本文模型效果最好

PDBbind数据集:
生物物理学任务
对于 PDBbind 数据集,本文模型 在基于图的模型中 取得了最好的性能,但并没有超过 基于网格的 特征提取方法。
可能对于配体-蛋白质结合的数据,最好的选择是建立一个网格来提取立体像素。
对于对接后的化合物,应该给出更多的信息,如配合物对接的对接位姿、能量和化学键等,而网格方法可以更好地描述配合物的3D结构。

baseline:
1、3、4是基于机器学习的;2、5、6、8是基于图的
①RF:使用多个决策树来训练和预测样本的分类器。它也可以用于回归问题。
②Muititask:多任务网络,是一个可以共享权重并处理多个任务的网络。
③XGBoost:梯度提升(XGBoost)是一种提升方法。,要思想是在前一个模型的损失梯度减小的方向上建立模型。 ④KPR:核脊回归,
是Ridge Regression(RR,脊回归)的kernel版本。(与支持向量回归类似。) ⑤DAG:有向无环图,使用RNN处理分子的
无向循环图。 ⑥GC:graph convolutional models,圆形指纹,直接使用卷积处理分子图 并 提取有效的分子表示。
⑦Weave:提出了一种GNN变体应用于 虚拟筛选 中分子表示 学习阶段 ⑧MPNN:消息传递神经网络,是一个通用框架。
⑨SGC:通过消除非线性和折叠连续层之间的权重矩阵,减少了GCN不必要的复杂性和冗余
⑩AGNN:基于注意力的图神经网络,去除GNN中所有全连接层,并使用 基于图的注意力层 代替 网络中的传播层,以便可以动态、自适应地学习。
①①GAT:通过叠加 mask的自注意层, 在卷积过程中 赋予不同节点 不同的重要性;并且可以同时处理 不同大小 的域。
①②ARMA:ARMA 是基于 自回归 移动 平均滤波器 的 图卷积层,其中滤波器具有 递归和分布式公式
3.5 分子指纹的影响
横轴是RMSE,纵轴是数据集
(1)在所有数据集上,单独使用指纹输入的模型表现不佳。这可能是由于 预定义的 分子特征信息不足 以及 特征与分子性质之间的相关性低,这使得模型无法提取潜在的有效信息。
(2)对于不同数据集,指纹对模型的贡献不同,这可能是由于提取的特征和分子性质之前的关系。
(在未来的工作中,可以通过选择合适的指纹特征来预测分子的不同特性,进一步提高模型的性能。)

4 结论
本文使用图结构来表示分子数据,将其输入CNN以发现每个原子之间的关系。其亮点如下:
①设计了一个 卷积空间图嵌入层(C-SGEL),使用一维卷积来处理分子中每个原子的空间图矩阵;然后,堆叠多个 卷积空间图嵌入层(C-SGEL)以构建卷积空间图嵌入网络(C-SGEN)。
②结合了分子指纹进行预测。
这篇关于Molecule Property Prediction based on Spatial Graph Embedding | 基于空间图嵌入的分子性质预测 | Graph | GNN的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!