本文主要是介绍PSP - 蛋白质复合物 AlphaFold2 Multimer MSA Pairing 逻辑与优化,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
欢迎关注我的CSDN:https://spike.blog.csdn.net/
本文地址:https://spike.blog.csdn.net/article/details/134144591
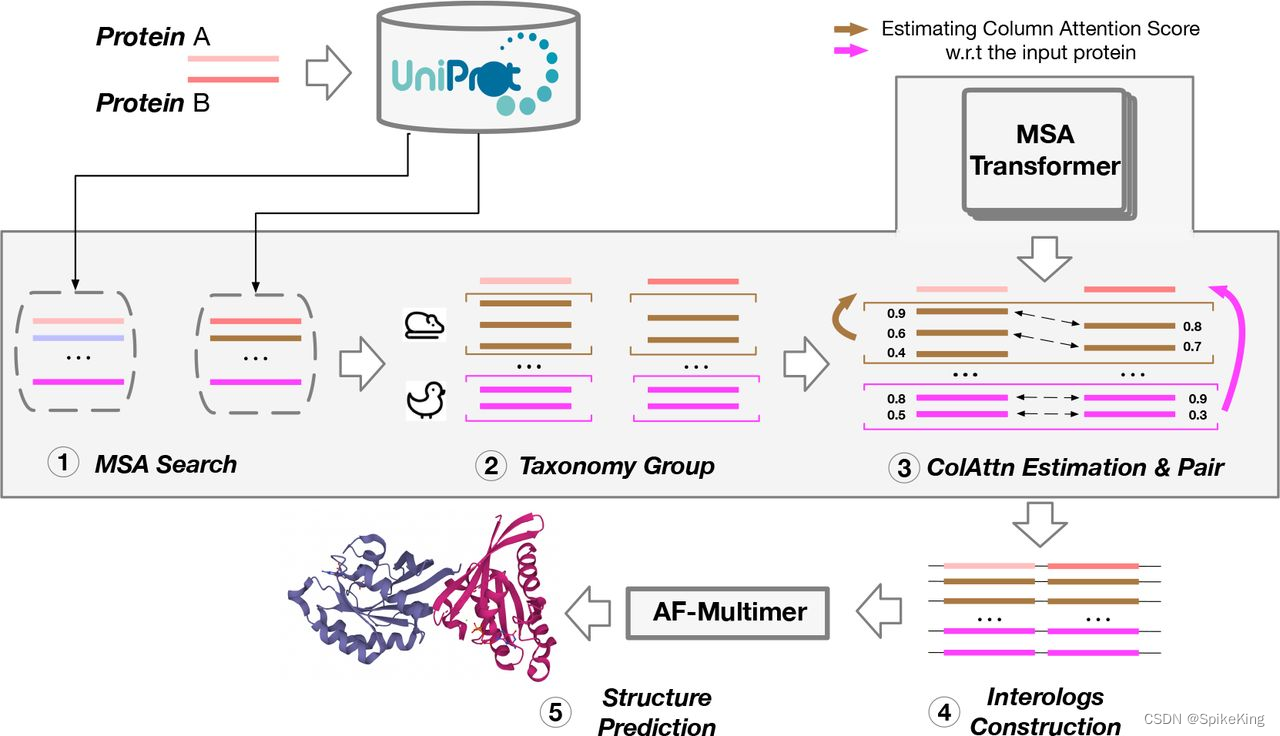
在蛋白质复合物结构预测中,当序列 (Sequence) 是异源多链时,无论是AB,还是AABB,都需要 MSA 配对,即 MSA Pairing。在 MSA 的搜索过程中,按照单链维度进行搜索,通过 MSA Pairing 进行合并,作为特征输入至 Multimer 结构预测。
控制 MSA 数量,包括需要 3 个超参数:
max_msa_crop_size,用于确定 MSA 的长度,默认设置 2048max_msa_clusters,用于确定推理中 MSA 特征的长度,默认设置 252max_extra_msa,用于限制推理中 Extra MSA 特征的长度,默认设置 1024
这 3 个参数,依次设定,从前到后相互包含,可以根据不同情况进行调节,其中 第1个参数 > (第2个参数 + 第3个参数)。
默认单链的搜索文件如下:
bfd_uniref_hits.a3m
mgnify_hits.sto
pdb_hits.sto
uniprot_hits.sto
uniref90_hits.sto
其中 uniref90_hits.sto 用于 MSA Pairing,pdb_hits.sto 用于 模版 (Template) 特征,bfd_uniref_hits.a3m、mgnify_hits.sto、uniref90_hits.sto,用于单链 MSA 特征。我们以 ABAB 格式的 4 链 PDB 进行假设。
优化1:MSA Pairing 默认只使用 uniprot_hits.sto,当数量较少时,可以使用 uniref90_hits.sto 作为补充。
源码 openfold/data/data_pipeline.py,如下:
# ++++++++++ 补充 MSA Pairing 源的逻辑 ++++++++++ #
# 标准的 AF2 Multimer 流程中没用 target_seq,即 target_seq 是 None
# logger.info(f"[CL] target_seq: {target_seq}")
msa = parsers.parse_stockholm(result, query_seq=target_seq)
msa = msa.truncate(max_seqs=self._max_uniprot_hits)msa_extra = parsers.parse_stockholm(result_extra, query_seq=target_seq)
msa_extra = msa_extra.truncate(max_seqs=self._max_uniprot_hits)logger.info(f"[CL] all_seq msa: {len(msa.sequences)}, add uniref msa: {len(msa_extra.sequences)}")
all_seq_features = make_msa_features([msa, msa_extra])
logger.info(f"[CL] all_seq msa: {all_seq_features['msa'].shape}")
# ++++++++++ 补充 MSA Pairing 源的逻辑 ++++++++++ #
优化2:当单链 MSA 数量较少时,使用 uniprot_hits.sto 作为 MSA 的补充。
源码 openfold/data/data_pipeline.py,如下:
# ++++++++++ 补充单链 MSA 序列的逻辑 ++++++++++ #
msa_seq_list = set()
for _, msa in msa_dict.items():for sequence_index, sequence in enumerate(msa.sequences):msa_seq_list.add(sequence)
msa_seq_list = list(msa_seq_list)
thr = 64 # 这影响没有 pairing 的序列,数值不宜过大
msa_size = len(msa_seq_list)
if msa_size < thr and uniprot_path:logger.info(f"[CL] single msa too small {msa_size} < {thr} (thr), uniprot_path: {uniprot_path}")with open(uniprot_path) as f:sto_string = f.read()msa_obj = parsers.parse_stockholm(sto_string)msa_seq_list += msa_obj.sequencesmsa_seq_list = list(set(msa_seq_list))diff_size = len(msa_seq_list) - msa_sizelogger.info(f"[CL] single msa from {msa_size} to {len(msa_seq_list)}, add {diff_size}")if diff_size > 0:msa_list.append(msa_obj) # 加入额外的数据
# ++++++++++ 补充单链 MSA 序列的逻辑 ++++++++++ #
优化3:当 MSA Pairing 数量过少时,尤其是 全链 Pairing 数量过少时,使用 其他物种 的 MSA 作为 MSA Pairing 的补充。
源码 openfold/data/msa_pairing.py,如下:
# ++++++++++ 补充 MSA Pairing 的逻辑 ++++++++++ #
thr = 128
num_all_pairing = len(tmp_dict1[num_examples])
if num_all_pairing < thr:logger.info(f"[CL] full msa pairing ({num_examples} chains) is too little ({num_all_pairing}<{thr}), "f"so add more!")tmp_dict2 = process_species(num_examples, common_species, all_chain_species_dict, prokaryotic, is_fake=True)# all_paired_msa_rows_dict = tmp_dict2tmp_item = list(tmp_dict1[num_examples]) + list(tmp_dict2[num_examples]) # 增补部分 MSAtmp_item = np.unique(tmp_item, axis=0) # 先去重tmp_item = tmp_item[:thr] # 再截取if len(tmp_item) > num_all_pairing:all_paired_msa_rows_dict[num_examples] = tmp_itemlogger.info(f"[CL] full msa pairing ({num_examples} chains) add to {len(tmp_item)}! ")
# ++++++++++ 补充 MSA Pairing 的逻辑 ++++++++++ #
假设序列是 AABB,顺序不重要,也可以是 ABAB,链式是 N c N_{c} Nc,MSA Pairing 只考虑 msa_all_seq 字段 (uniprot_hits 和 uniref90_hits 优化),即,A 链包括 MSA 数量是 L A L_{A} LA,B 链包括 MSA 数量是 L B L_{B} LB,MSA Pairing 数量是 L P a b L_{P_{ab}} LPab 。其中 MSA Pairing 包括 2 至 N c N_{c} Nc 个,例如 4 链,就是可以 Pairing 成2链、3链、4链等 4 种情况,只有 1 链时,被抛弃。
源码 openfold/data/msa_pairing.py,即:
# Skip species that are present in only one chain.
if species_dfs_present <= 1:continue
在 MSA Pairing 的过程中,修改 msa_all_seq 字段的 MSA 顺序,同时去除 只有 1 链 (没有配对) 的情况,假设最终 MSA Pairing 的数量是 L P a b L_{P_{ab}} LPab,全部链都是相同的,填补空位。
通过 msa_pairing.merge_chain_features() 函数,将单链 MSA 的合并至一起,即 bfd_uniref_hits.a3m、mgnify_hits.sto、uniref90_hits.sto 的全部 MSA,组成 msa 字段特征。其中 MSA 参数1 即 max_msa_crop_size,表示合并 MSA 的最大数量。例如 链 A 的 msa_all_seq 数量是 900,最大是 2048,则 单链 msa 字段的数量最多是 1148,其余随机舍弃,即1148+900=2048。
源码 openfold/data/msa_pairing.py,注意 feat_all_seq 在前,feat 在后,即 MSA Pairing 更重要,即:
def _concatenate_paired_and_unpaired_features(example: pipeline.FeatureDict,
) -> pipeline.FeatureDict:"""Merges paired and block-diagonalised features."""features = MSA_FEATURESfor feature_name in features:if feature_name in example:feat = example[feature_name]feat_all_seq = example[feature_name + "_all_seq"]merged_feat = np.concatenate([feat_all_seq, feat], axis=0)example[feature_name] = merged_featexample["num_alignments"] = np.array(example["msa"].shape[0], dtype=np.int32)return example
通过 openfold/data/data_transforms_multimer.py 函数,将输入的 msa 特征 (合并 msa 和 msa_all_seq) 进行截取,先截取 max_seq,再截取 max_extra_msa_seq,即第 2 个和第 3 个参数,max_msa_clusters 和 max_extra_msa,作为最终的训练或推理 msa 特征。
logits += cluster_bias_mask * inf
index_order = gumbel_argsort_sample_idx(logits, generator=g)
logger.info(f"[CL] truly use msa raw size: {len(index_order)}, msa: {max_seq}, extra_msa: {max_extra_msa_seq}")
sel_idx = index_order[:max_seq]
extra_idx = index_order[max_seq:][:max_extra_msa_seq]for k in ["msa", "deletion_matrix", "msa_mask", "bert_mask"]:if k in batch:batch["extra_" + k] = batch[k][extra_idx]batch[k] = batch[k][sel_idx]
通过不同的训练模型,与不同的参数,进行蛋白质复合物的结构预测。
这篇关于PSP - 蛋白质复合物 AlphaFold2 Multimer MSA Pairing 逻辑与优化的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!


