本文主要是介绍Nature子刊:教你零基础开展微生物组数据分析和可视化,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!

使用MicrobiomeAnalyst进行微生物组数据的全面统计、功能和元分析
Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data
Nature Protocols
Impact Factor 11.334
https://doi.org/10.1038/s41596-019-0264-1
发表日期:2020-01-15
第一作者:Jasmine Chong
通讯作者:Jianguo Xia(夏建国,jeff.xia@mcgill.ca)
合作作者:Peng Liu, Guangyan Zhou
主要单位:
麦吉尔大学寄生虫研究所,加拿大魁北克省圣安妮德贝勒维 (Institute of Parasitology, McGill University, Ste-Anne-de-Bellevue, Quebec, Canada)
摘要
MicrobiomeAnalyst是一个方便易用的网页工具,是系统全面分析微生物组学数据的分析网站。目的是为未经生物信息学系统培训的研究人员和临床医生能参考目前主流分析方法轻松挖掘微生物组学数据,包括数据预处理,统计分析,功能分析和与公开数据集或已知微生物特征进行比较。该平台目前包含标记基因数据分析 (Marker-gene Data Profiling ,MDP)、鸟枪数据分析 (Shotgun Data Profiling ,SDP)、公共数据投影 (Projection with Public Data ,PPD)和分类集群富集分析 (Taxon Set Enrichment Analysis ,TSEA)四个模块;MDP和SDP负责分析标记基因和鸟枪法宏基因组及转录组数据,PPD和TSEA负责可视化比较或者关联用户数据和公共数据库数据。一次完整的分析最快 70 分钟内完成,视数据量大小而定;下文中将描述详细的使用步骤介绍。
背景Introduction
高通量测序技术的快速发展改变了各种环境的微生物群落的研究。本文的“微生物组”(microbiome) 是指定居于特定生物生态位的微生物,包括其基因组含量和代谢产物。现在普遍认为微生物群与宿主息息相关,如果微生物群的生态系统失衡将对宿主不利。目前研究微生物组学的主要方法有:(i) 标记基因组学,以获得群落微生物的概貌。(ii) 鸟枪法宏基因组学,以了解微生物组的功能潜力,(ii) 宏转录组学,通过基因表达谱来测量其功能活性,目前几个能通过处理原始下机数据得到特征丰度表,例如:QIIME,mothur,UPARSE, DADA2,One Codex,Kraken,MetaPhlAn;特征丰度表和样本相关信息(元数据)是下游统计分析和功能解释的关键。
目前微生物组学数据处理有以下几个关键的挑战:
-
每个样品的测序数据量(即文库大小)差异大,需要先对数据进行适当的归一化,然后才能进行有意义的统计分析;
-
丰度表中最低分类水平值非常稀疏,这种稀疏性可能是由于采样不足或实际没有该分类单元而引起的。
-
微生物组数据是组成型的。即如果优势特征相对增加,那其他特征的相对丰度(比例)将减小,即使它们的绝对丰度保持恒定。
正由于微生物组数据具有这些特征,在处理数据时应该加以考虑,正确处理,目前,R 的 phyloseq 包提供了丰富的功能来处理特征表,分类树和元数据处理。但是基于编码的界面对于临床工作人员等科研人员十分不利。
MicrobiomeAnalyst这是应对这种情况而开发的工具。因此为无需专业编程技能就能可轻松进行微生物组数据的系统综合统计分析,交互式可视化和meta分析。用户可以从多种完善的方法中进行选择,并实时浏览结果,以更好地了解其数据。自2017年首次发表以来,MicrobiomeAnalyst已逐渐在微生物组研究人员中流行。在过去的12个月中,该Web服务器已处理了来自全球20,000多个用户的70,000份数据分析作业。我们一直在积极改进当前功能,并根据用户的反馈和文件的发展添加新功能。为了满足不断增长的用户流量和计算需求,服务器最近已迁移到高性能Google Cloud平台。
分析流程和界面设计
Overview of the analysis workflow and the interface design
MicrobiomeAnalyst 的总体工作流程如图 1 所示。共有四个模块:标记基因/扩增子数据(Marker-gene Data Profiling, MDP)、宏基因组数据(Shotgun Data Profiling, SDP)、综合公共数据(Projection to Public Data, PPD)、分类单元集富集分析(Taxon Set Enrichment Analysis, TSEA)。四个模块共享相同的常规工作流程-数据准备,数据分析和可视化探索。在数据准备阶段,上载用户数据以进行过滤和标签化。此后,可以对处理后的数据执行各种统计和可视化方法,以检测例如总体模式、重要功能、潜在的交互作用和功能见解。对于MDP模块,总共提供了19种精心选择的方法(图2)。每种方法的Web界面允许用户调整关键参数,以进行交互式分析和结果的可视化探索。经过基本数据预处理,就数据类别进行对应的下游分析,可通过交互式对核心参数调整。
图1 MicrobiomeAnalyst工作流程概述。

图1 | MicrobiomeAnalyst工作流程概述。MicrobiomeAnalyst包含四个模块:标记基因数据分析(MDP),鸟枪测序分析(SDP),公共数据元分析(PPD)和富集分析(TSEA)。在他们各自的流程里阐明了每个模块的关键功能。PC,主坐标。
Fig. 1 | Overview of the MicrobiomeAnalyst workflow. MicrobiomeAnalyst comprises four modules: Marker-gene Data Profiling (MDP), Shotgun Data Profiling (SDP), Projection with Public Data (PPD), and Taxon Set Enrichment Analysis (TSEA). The key functions of each module are illustrated in their respective boxes. PC, principal coordinate.
图2 全面的数据分析和报告生成
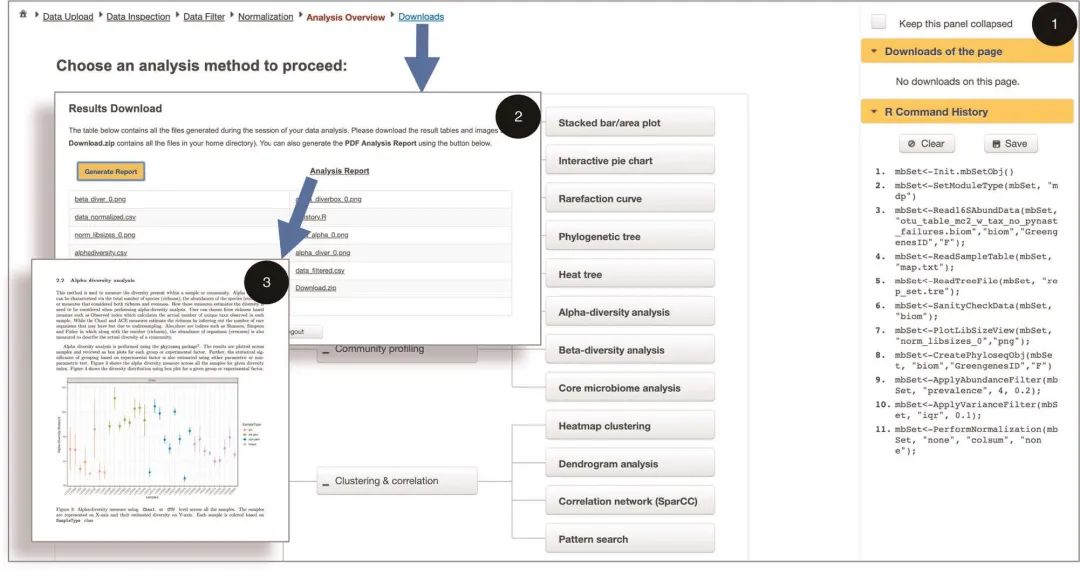
图2 | 全面的数据分析和报告生成。扩增子MDP“分析概述”页面(1)的屏幕截图,展示了可用的分析方法集和。左上角显示导航栏,当前页面以红色突出显示。页面右侧的“ R命令历史记录”面板显示所有可在R 中分析的R命令。“页面下载”面板显示从当前页面生成的结果。用户还可以点击顶部导航栏中的“下载”链接,进入“结果下载”页面(2)并批量下载所有结果,并生成全面的分析报告(3)。
Fig. 2 | Comprehensive data analysis and report generation. A screenshot of the MDP ‘Analysis Overview’ page (1) to illustrate the comprehensive set of analysis methods available. The top left corner shows the navigation track with the current page highlighted in red. The ‘R Command History’ panel to the right of the page displays all underlying R commands. The ‘Downloads of the page’ panel displays the results generated from the current page. Users can also click the ‘Downloads’ link from the top navigation track to enter the ‘Results Download’ page (2) and batch-download all results as well as to generate a comprehensive analysis report (3).
MicrobiomeAnalyst 还提供了动态导航轨道和实时系统消息来指导用户完成数据准备和分析的每个步骤(图2)。并在右侧提供“结果下载”下载该页的分析结果,和“R命令历史记录”面板,该面板显示实时发生的底层 R 命令,以帮助提高微生物组数据分析的透明度,灵活性和可重复性。用户可以从GitHub(https://github.com/xia-lab/MicrobiomeAnalystR)安装基础的R包(MicrobiomeAnalystR),并使用这些R命令在本地重现其结果。最近添加了此功能,以遵循与我们的MetaboAnalyst Web服务器及其配套的MetaboAnalystR包相同的概念,帮助提高微生物组数据分析的透明度,灵活性和可重复性。
与其他网页工具比较
Comparison with other web-based tools
目前主流的微生物数据处理的网站有MG-RAST、VAMPS、Calypso。MG-RAST:用于注释和存储原始宏基因组学数据的公共资源,提供基础的统计分析和可视化,高级分析推荐 matR 包处理; VAMPS:主要提供可视化分析,如热图,饼图和主坐标分析 (PCoA) 图等; Calypso:支持数据处理以及微生物组数据的多样性,比较和网络分析。与这些工具相比,MicrobiomeAnalyst 实时可见分析过程及其具体R命令以提高透明度和可重复性,可轻松导航到指定数据处理步骤;胜任复杂的分析任务。例如,MDP模块当前提供了19种经过精心选择的统计分析和可视化方法。物种富集分析是 MicrobiomeAnalyst 独有的功能;其次, MicrobiomeAnalyst的用户高度评价的另一个功能是在整个数据分析过程中创建的可用于发布出版物级别的图形输出。MicrobiomeAnalyst通过提供全面的分析报告和R命令历史记录以及其配套的R包提高了数据分析的透明度和可重复性。表1显示了MicrobiomeAnalyst与这三个基于Web的工具之间的详细比较。
表1 比较MicrobiomeAnalyst和其他三种网页工具对微生物组数据分析的流程
Table 1 | Comparisons of MicrobiomeAnalyst with other web-based tools for microbiome data analysis

局限性
Limitations
但是,MicrobiomeAnalyst 不能处理原始测序数据,由于网络和服务器成本等问题,不提供处理原始数据的功能,而专注于实时交互式数据分析,但是也提供了MicrobiomeAnalyst R 包,用户可用该包自己线下整理好丰度表再上传分析。MicrobiomeAnalyst 目前只能处理不同处理的数据,对于时间序列数据不行,时序数据正在研发。目前 MicrobiomeAnalyst 每次打开新的会话时,用户都需要重新上传并重新执行数据处理步骤。这可能会影响某些分析结果的可重复性,例如“随机森林”的分类结果或网络分析(SparCC) 的校正 P 值,正在开发允许注册用户保存其工作进度和历史并在以后的某个时间恢复历史分析,继续开展分析。
实验设计
Experimental design
下面的实验方法分为四个部分,以展示MicrobiomeAnalyst中的所有四个模块:(i)对16S rRNA标记基因丰度数据的全面分析(步骤1至30);(ii)进行预测性功能分析,然后进行《京都基因与基因组百科全书》(KEGG)直系同源(KO)丰度表的途径富集分析和网络可视化(步骤31-49);(iii)使用公共数据集进行视觉数据探索(步骤50-56);(iv)分类单元集富集分析(步骤57-63)。下面的过程中提供了详细的分步教程。
综合分析16S rRNA丰度数据
Comprehensive analysis of 16S rRNA abundance data
MDP模块是使用最频繁的模块,包含MicrobiomeAnalyst当前可用的所有方法的一半以上。通常,微生物组数据分析的第一个问题是确定数据内是否有任何模式。这种探索性分析是通过常用的生态方法进行的,包括α和β多样性分析。然后可以使用多元统计信息来评估此类模式的鲁棒性。下一步的逻辑步骤是确定哪个分类单元负责观察到的差异。重要分类单元及其相关性或共现模式的识别可以使用不同的单变量统计方法或更复杂的多变量程序来完成。对于经过深入研究的微生物群落,例如人类肠道微生物群,也有可能预测其功能潜力。由此产生的基因丰度数据可以提供重要的功能见解,而无需执行鸟枪法宏基因组测序。
基因丰度数据的功能分析和网络可视化
Functional profiling and network visualization of gene abundance data
SDP模块提供了一组相似的方法,用于模式发现和对由预测功能分析或宏基因组学/宏转录组学产生的基因丰度数据进行比较分析。SDP的独特功能是其基于模块,途径和代谢网络的功能注释。MicrobiomeAnalyst使用户可以轻松地可视化这些功能在样本和研究条件中的分布。它还支持显式统计检验以识别丰富的功能。用户可以在新陈代谢网络环境中以交互方式浏览结果,以进一步了解功能。
与公共数据集的可视化比较
Visual comparison with a public dataset
随着公共数据集数量的增加,荟萃分析已成为比较和假设产生的强大方法。PPD模块旨在使用户能够在兼容的公共数据集范围内直观地浏览自己的16S rRNA数据。这些公共数据集主要来自Qiita。用户选择用于荟萃分析的数据集必须共享至少20%的分类学特征才能进行有意义的比较。在此模块中,将对用户和公共数据进行共同处理,然后共同投影到交互式3D PCoA图中以进行视觉比较。用户可以比较样品的分类组成,以找出哪些分类单元在推动组分离。这使用户能够将其数据关联到上下文中以获得全局视角,以便例如识别不同环境或人群之间的成分差异。
分类群列表的富集分析
Enrichment analysis of a list of taxa
经过比较分析,用户将产生与感兴趣的表型显著相关的分类单元列表。但是,这样的列表通常缺乏用于发展假设或获得机制洞察力的环境。富集分析是一种已经流行的用于解释基因和代谢物列表的方法,可以用于从分类群列表中获得更深入的见解。但是,一个关键障碍是需要创建一个与基因组或代谢物组相似的分类单元集的全面而有意义的集合。为解决这一差距,我们从微生物组研究的不同领域的高影响力期刊(影响因子> 3)中手动选择了2,393个分类单元。可以从MicrobiomeAnalyst网站的“资源”页面下载这些分类单元集。这些分类单元集进一步分为五类:与(i)宿主单核苷酸多态性(SNP),(ii)宿主内在因素(例如疾病),(iii)宿主外在因素(例如饮食)相关的分类单元集和生活方式),(iv)环境因素(例如化学暴露)和(v)微生物固有因素(例如流动性和形状)。
框1 16S扩增子数据预处理过程
Box 1 | Preprocessing of raw 16S rRNA amplicon sequencing data

此框描述了原始序列数据预处理的一般步骤和可用工具。
标记基因的扩增子测序是一种广泛用于跨不同宿主和环境的微生物群落分类学分析的方法。从测序平台获得原始序列后,需要生物信息学流程将原始读取转换为分类信息。传统上,原始读取会转换为OTU,即满足97%相似性阈值划归为一个OTU。现在通常建议将原始读数转换为高分辨率的ASV,可以根据其独特的生物学序列对其进行鉴定,以促进整个研究的荟萃分析。所有生物信息学流程的主要预处理步骤是(i)测序序列的质量控制,(ii)序列的聚类和(iii)分类分配。常用的管道包括QIIME,mothur,UPARSE,以及最近的DADA2。DADA2的工作原理是生成一个参数错误模型,该模型将对所有原始测序数据进行训练,并应用该模型将序列错误纠正和合并为ASV。MicrobiomeAnalystR软件包集成了DADA2,可用于原始16S rRNA扩增子测序数据。
材料Materials
设备Equipment
电脑要求Computer requirements
-
浏览器要求:MicrobiomeAnalyst可在所有主流Web浏览器上运行。为了获得最佳体验,我们建议使用Google Chrome v.75 +,Firefox v.67 +,Safari v.12 +或Microsoft Internet Explorer v.11 +。必须在浏览器中启用JavaScript。
-
Internet连接要求:强烈建议具有宽带连接。
-
硬件要求:内存RAM > 2 GB,并且屏幕分辨率至少为1200×800。
数据文件Data files
-
输入文件(Input files)。MicrobiomeAnalyst的主要输入文件是三个制表符分隔的纯文本文件:一个特征丰度表,其中包含多个样本中特征(操作分类单位(OTU)/ ASV /基因)的读长计数,这些特征的分类文件(OTU / ASV)和描述这些样本的组信息的元数据文件。MicrobiomeAnalyst还接受QIIME流程生成的BIOM文件以及mothur流程的输出。另外,如果用户希望执行系统发育树分析或基于UniFrac距离的分析,则需要使用任何常用算法生成的树文件。有关这些文件格式的更多详细信息,请参见框Box 2。
-
示例数据集(Example datasets)。MicrobiomeAnalyst提供了多个示例数据集以进行测试。在每个模块的数据上传页面上,用户可以直接使用“示例数据集进行测试”面板中的示例数据。此协议中使用了三个示例数据集。第一个数据集由来自小儿炎症性肠病(IBD)患者和从整合人类微生物组计划(iHMP)获得的健康对照的43个粪便样本组成。这些数据是使用MicrobiomeAnalystR软件包中集成的DADA2流程进行预处理的。这些数据将用于MDP和TSEA模块,以探索两组之间的微生物差异。第二个数据集由21个粪便微生物组样本组成,这些样本来自对衰老小鼠的研究。这些数据将首先由MDP模块使用,以生成预测的基因丰度表,然后将其用作SDP模块的输入。第三个数据集由来自北美和南美耕地的26个环境微生物组样本组成。该数据集旨在与PPD模块一起使用,以与其他微生物组数据集进行荟萃分析。
设置部署Equipment setup
下载示例数据 Download the example data
转到MicrobiomeAnalyst主页(https://www.microbiomeanalyst.ca),然后从顶部菜单栏中单击“资源Resources”。在“示例数据集Example Datasets”选项卡上,单击每个压缩的文件夹以将其保存在计算机上。下载它们之后,解压缩每个文件夹,以便可以访问所有文件以上传到MicrobiomeAnalyst。
框2 数据格式化和上传
Box 2 | Data formatting and upload

此框说明如何为MicrobiomeAnalyst准备处理后的微生物组数据。
MicrobiomeAnalyst接受从几个常用的生物信息学流程生成的丰度数据。这些文件可以以纯文本格式(.txt或.csv)上传,也可以直接作为.biom或.shared文件上传。用户还必须提供描述相同样品的组信息的分组信息文件。以下是有关如何格式化MicrobiomeAnalyst的丰度,分类和分组信息文件的简短说明。
-
微生物丰度文件(.txt / .csv)
丰度表应设置格式,以使特征于行中,样本位于列中。第一行应以“ #NAME”开头。如果特征名称包含微生物分类单元名称,请确保用使用分隔符;分隔(例如, Bacteria; Firmicutes; Clostridia )。如果特征不包含特定的分类名称(例如,OTU000001),则还必须提供分类注释文件(请参见下文)。
-
分类文件(.txt / .csv)
分类文件的格式应设置为使特征名称在第一列中,表开头必须是‘#TAXONOMY ’。应在“ Phylum”,“ Class”,“ Order”,“ Family”,“ Genus”和“ Species”列的下包含所有功特征的分类信息。特征名称必须与出现在丰富文件中的特征名称匹配。
-
分组数据文件(.txt / .csv)
分组数据文件应设置格式,以便第一列包含样本名称,命名为“ #NAME”。随后的列包含有关组或其他实验因素信息。样品名称必须与丰度文件中显示的样本名称匹配。
程序 Procedure
阶段1:全面分析16S丰度数据
Stage 1: Comprehensive analysis of 16S abundance data
大约30分钟,具体取决于数据集的大小
-
启动。转到MicrobiomeAnalyst主页(https://www.microbiomeanalyst.ca),然后单击“标记数据分析(MDP)”圆圈以进入MDP模块。故障排除建议详见**表2**。
-
上传数据。在框2中可以找到准备输入文件的详细说明。进入MDP模块后,单击“用于测试的示例数据集Example data sets for testing”以展开包含所有可用示例数据集的面板。选择“数据类型Data Type”下列出的“Pediatric IBD”数据集。点击“提交”按钮上传数据(稍等进入下一页数据概述)。或者,选择“纯文本表格格式”面板。点击“ OTU / ASV表”旁边的“选择文件”按钮,然后找到“ ibd_asv_table.csv”文件。对“ ibd_meta.csv”,“ ibd_taxa.txt”和“ ibd_tree.tre”文件重复此步骤。在“分类标签Taxonomy labels”下拉菜单中,单击“Greengenes Taxonomy”。点击“提交Submit”按钮上传数据。
-
数据完整性检查。此页面包含两个选项卡。第一个标签为“文本摘要Text Summary”,提供上传文件的文本摘要。第二个标签“库大小概述Library Size Overview”以图形方式描述了所有上传样本的读长计数(用于下游抽平准备化选择参数),这对下游数据过滤和规范化很有用。点击页面底部的“继续”前进。
-
数据过滤。通常建议进行过滤以删除低质量的特征,从而改善下游统计分析。保留“低计数过滤器Low count filter”和“低差异过滤器Low variance filter”滑块的默认选择,然后单击“提交”以执行数据过滤。右上角将出现一条消息,指示数据过滤步骤的结果。请注意,过滤后的数据将不会用于alpha多样性分析,并且用户可以通过将相应的滑块拖动到零值来关闭过滤器。有关数据过滤的更多详细信息,请参见框3。单击页面右下方的“继续”,导航至下一页。
-
数据标准化。在“数据标准化Data Normalization”页面上,用户可以执行数据稀疏rarefying,缩放scaling和转换transformation。数据标准化的目的是对数据进行标准化以实现准确的比较。在框3中可以找到更多详细信息。保留选项的默认选择(仅将“数据缩放”设置为“总体缩放TSS”),然后单击“提交Submit”,然后单击“继续Proceed”以移至“分析概述”页面。
-
群落分析。用户可以使用“Alpha-diversity”和“ Beta-diversity”分析选项来评估微生物群落的多样性概况(更多信息请参见框4)。首先,请点击“分析概述Analysis Overview”页面中的“Alpha-diversity analysis”。
-
Alpha多样性。页面顶部是几个下拉菜单,用户可以在其中浏览不同的alpha多样性测度或选择分类级别以评估多样性差异。默认情况下,使用Chao1在特征(OTU / ASV)级别评估alpha多样性,并使用t检验评估显著性差异。页面的下半部分包含结果的两个图形摘要。左侧是点图,显示了样本之间的alpha多样性度量,而右侧是框图,总结了各个组的alpha多样性度量。从这些结果可以看出,小儿IBD患者和健康对照组的样本内多样性存在显着差异:与对照组相比,IBD患者的α多样性测量值明显更低。
-
(可选)探索不同的Alpha多样性衡量标准;每个人对群落结构都做出不同的假设,因此将揭示群落结构的不同方面(更多详细信息,请参见框4)。还可以尝试使用不同的分类标准,以查看在更高的分类标准中是否可以观察到相同的趋势。
-
Beta多样性。点击页面顶部导航栏中的“分析概述Analysis Overview”链接。接下来,点击“Beta Diversity analysis”。此页面的上半部分包含用于Beta多样性分析的参数(更多详细信息,请参见框4)。页面底部的两个选项卡分别显示2D和3D PCoA图。默认情况下,使用Bray-Curtis指数评估小儿IBD患者和对照组之间的多样性差异。变异的多元方差分析(PERMANOVA)表明,两组的聚类差异显著(P值<0.001)。
-
3D PCoA探索。单击“Interactive PCoA 3D”标签,以基于前三个主坐标轴的交互式3D散点图进一步浏览PCoA结果(图3)。使用鼠标旋转并放大和缩小图。同样,我们发现两组之间有明显的分隔。
图3 用于beta多样性分析的交互式3D PCoA图
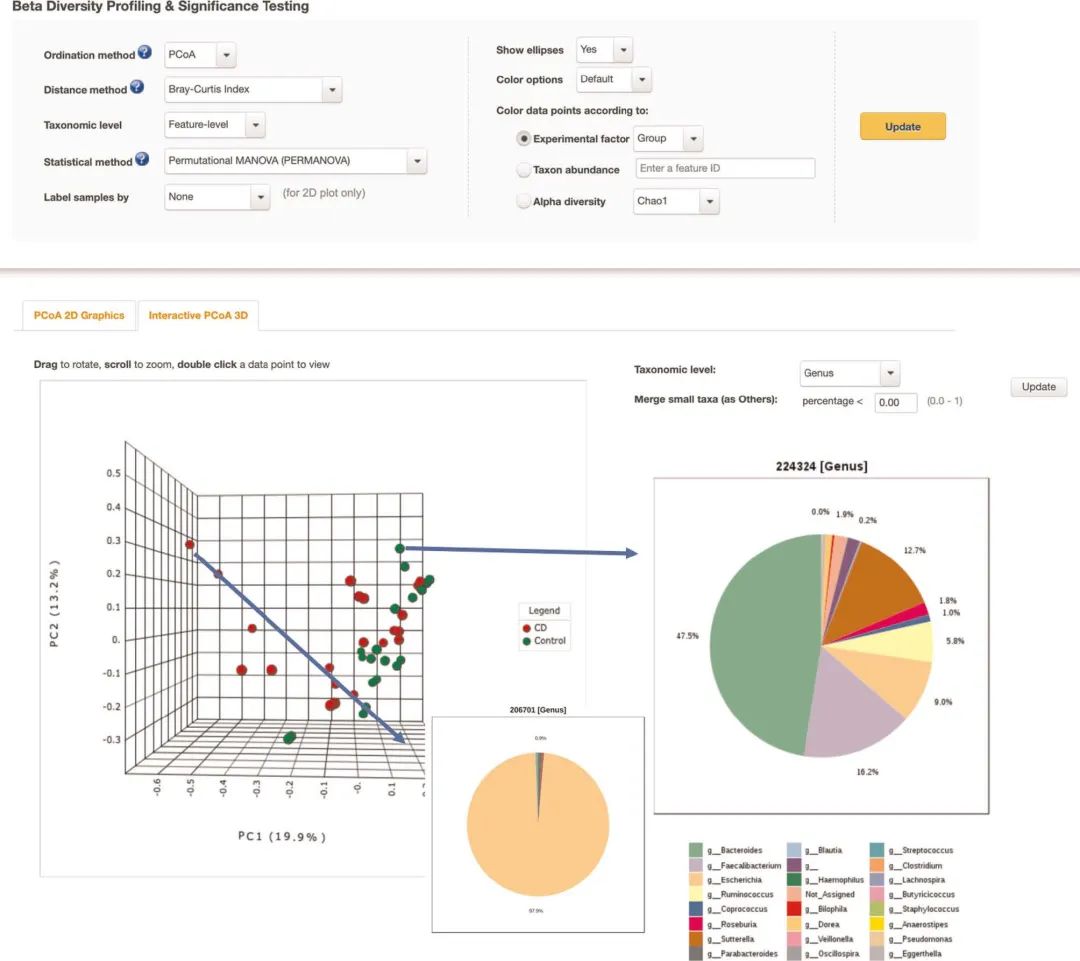
图3 | 用于beta多样性分析的交互式3D PCoA图。Beta多样性分析生成的3D PCoA图和饼图的屏幕快照。用户可以旋转图形或双击任何样本,以在选定的分类学级别通过饼图查看其微生物丰度信息。显示了两个饼图,一个来自对照样品,另一个来自克罗恩病人(CD)样品。对照样品以拟杆菌属为主导,而CD样品以大肠埃希菌为主导。
Fig. 3 | Interactive 3D PCoA plot for beta-diversity analysis. A screenshot of the 3D PCoA plot and pie charts generated by the beta-diversity analysis. Users can rotate the graph or double-click any sample to view a pie-chart summary of its microbial abundances at a selected taxonomic level. Two pie charts, one from a control sample and one from a Crohn’s disease (CD) sample, are shown. The control sample is dominated by Bacteroides, whereas the CD sample is dominated by Escherichia.
-
双击IBD组(红色)中的几个数据点(代表样本),以查看该样本的分类学丰度的相应饼图。用户可以更改饼图的分类标准级别,也可以合并小的(在用户指定的临界值以下)分类单元。将分类级别更改为“属Genus”,然后单击“更新Update”。
-
双击几个对照样品并查看其相应的饼图。注意健康对照组和IBD患者之间的分类差异。例如,以第一主轴左右两个点为例,似乎来自IBD患者的样品主要由大肠杆菌组成,而健康对照则由拟杆菌属组成(图3)。
-
(可选)默认情况下,在Bray-Curtis差异指数上使用PCoA可视化beta多样性分析,并使用PERMANOVA进行评估。为了获得不同的观点,请将排序方法更改为“非度量多维标度(NMDS)”,将统计方法更改为“相似性分析(ANOSIM)”,这两种方法都是基于排序列的方法。然后将距离方法更改为“Unweighted UniFrac Distance”,该方法使用要素之间的系统发育距离,而不是要素的丰度信息(有关更多详细信息,请参见框4)。点击“更新”并浏览结果。我们看到组间仍然有显著差异,但没有PCoA Bray-Curtis距离下明显,因为NMDS采用秩排序,而且采用无权重方法去除了丰度信息,从不同角度会看到不同的结果。
-
热树分析。返回“Analysis Overview”页面,然后单击“热树Heat Tree”。热树分析使用分类学分类的层次结构来描述微生物群落的分组的相对丰度。该页面的上部包含用于创建和自定义热树的关键参数。将“属Genus”设置为当前分类标准,为热树布局(Heat tree layout)指定“ Reingold-Tilford”,将“比较Comparison”保留为当前查看模式,然后选择“ CD_vs_Control”进行感兴趣的比较(CD_vs_Control) 。单击“提交”以生成相应的热量树(图4)。由于算法的随机性,树的布局可能会略有变化。可以从页面的右上方面板下载差异表,该差异表包含使用不同分类级别的非参数Wilcoxon检验进行的组间比较。
图4 树状热图的分类差异可视化
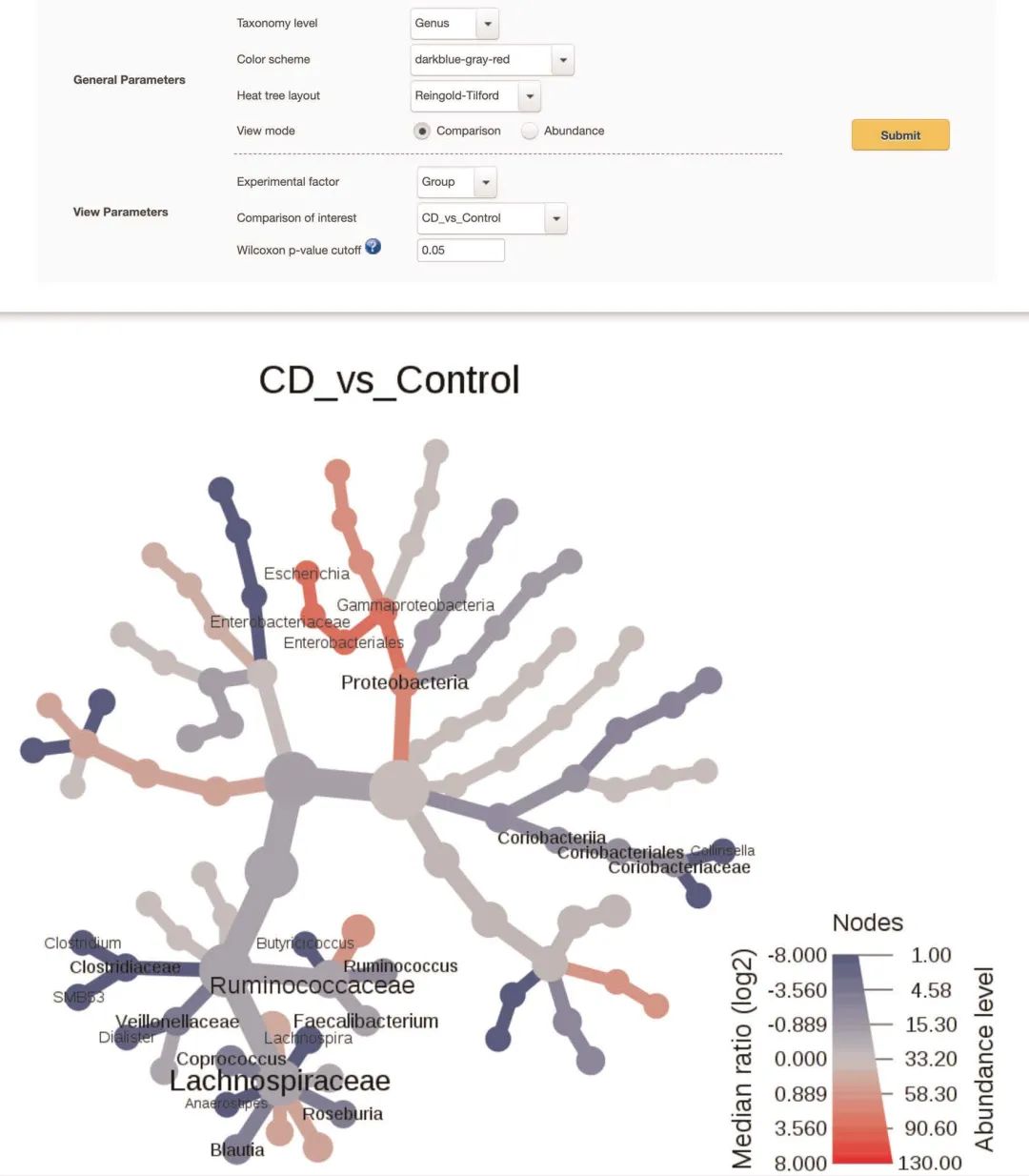
图4 | 树形热图的分类差异可视化。树形热图的屏幕截图,用于说明两个选定组之间的分类差异。页面顶部显示比较的两组,注意只能是两组之间比对。颜色渐变以及节点,边缘和标签的大小基于中位数丰度的log2比值。在这种情况下,蓝色和红色表示与对照组相比,克罗恩病患者的相应微生物分类单元分别较低和较高的程度。
Fig. 4 | Heat tree visualization of taxonomic differences. A screenshot of a heat tree to illustrate the taxonomic differences between the two selected groups. The top of the page shows the key parameters. The color gradient and the size of node, edge, and label are based on the log2 ratio of median abundance. In this case, blue and red indicate that corresponding taxa are lower and higher, respectively, in Crohn’s disease patients as compared with controls.
-
相关网络分析(Correlation network analysis)。点击顶部导航栏中的“Analysis Overview”,然后选择“Correlation network(SparCC)”。相关网络分析使用四种方法来计算分类特征之间的成对相关:SparCC,Pearson相关,Spearman秩相关和Kendall tau相关。尤其是,SparCC旨在解决由于微生物组数据的组成特性引起的虚假相关性问题(更多详细信息,请参见框5)。该页面的顶部包含执行相关分析和生成网络的所有必要参数。首先,请确保从“算法Algorithm”下拉菜单中选择了“ SparCC”。从“分类级别Taxonomy level”下拉菜单中选择“属Genus”,然后从“感兴趣的比较Comparison of interest”下拉菜单中选择“ CD vs Control”。保留P值和相关性的默认阈值。点击“Submit”以生成网络(图5)。当数据集中有许多特征和样本时,此步骤可能会很耗时。
图5 相关网络分析
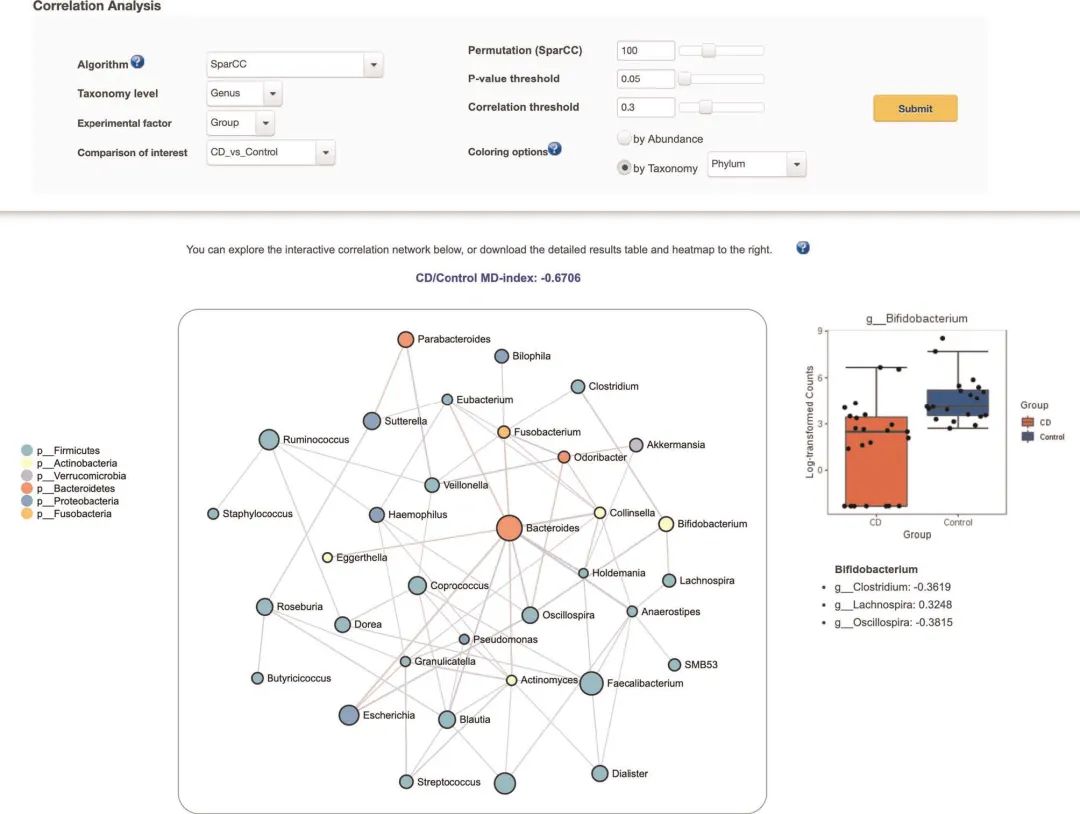
图5 | 相关网络分析。使用SparCC算法生成的相关网络的屏幕截图。图像的中心是相关网络,其节点表示属级别的分类单元,边表示分类对之间的相关性。节点根据门水平分类进行着色。右侧是双歧杆菌的箱形图,显示与健康对照组相比,CD患者的该菌丰度降低了。
Fig. 5 | Correlation network analysis. A screenshot of the correlation network generated using the SparCC algorithm. In the center of the image is the correlation network, with nodes representing taxa at the genus level, and edges representing correlations between taxa pairs. The nodes are colored on the basis of phylum. To the right is a box plot of Bifidobacterium showing reduced abundance in CD patients versus healthy controls.
-
探索相关网络(Exploring the correlation network)。在结果相关网络中,节点代表分类学特征,边代表大于两类分类之间的相关阈值。默认情况下,节点根据其丰富程度进行着色,并且边的宽度反映了分类单元之间相关性的强度。要更新节点的颜色,请选择“按分类法”,将分类法级别保持为“ Phylum”,然后单击“提交”(图5)。现在,根据节点的种类为节点着色,并在网络左侧添加颜色图例。网络也是互动的。例如,双击黄色节点Bifidobacterium(在左侧)。网络右侧将出现一个比较该分类单元丰度的箱形图,下面的数值表示该节点与其最邻近节点之间的相关系数。最后,网络顶部是MD指数(microbial dysbiosis index,微生物异常指数),它是对微生物组内部异常程度的经验估计。此处的MD指数为-0.67(该值在不同的运行中可能会略有不同),这表明与对照组相比,CD患者的分类单元丰富度总体下降了。
-
(可选)使用其他相关算法(Spearman算法,Pearson算法或Kendall算法;框5)比较相关分析的结果。
-
经典单变量分析。返回“分析概述”页面,然后点击“经典单变量分析Classical univariate analysis”。MicrobiomeAnalyst提供t检验/ ANOVA及其非参数对等检验。所有差分丰度分析的结果都遵循相同的布局。页面的上半部分包含用户可以用来自定义分析的参数,例如分类级别,统计方法和显著性临界值。页面的下半部分包含用于分析的结果表。表格中的要素按假阳性率(FDR)调整后的P值排名,而低于阈值的要素以橙色突出显示。
-
点击结果表“查看View”列下的“详细信息Details”链接。箱形图将出现在弹出对话框中,显示选定特征在不同组中的丰度。
-
(可选)探索在不同分类组别下确定的重要特征。
-
使用开发用于RNA-Seq数据分析的方法鉴定重要特征。在导航栏中单击“分析概述”以返回到“分析选项”页面。
-
点击“比较和分类Comparison & classification”选项中的“RNA-seq methods”。默认情况下,edgeR在算法(Algorithm)级别执行。与经典单变量分析相比,edgeR可识别56个重要特征(比一般非参检验更敏感,适合差异小的组间或找到更多差异,避免假阴性)。将分类级别更改为“种 Species”,然后单击“提交Submit”。总共鉴定出14种(切换为DESeq2只找到2种)。
-
顶部特征之一是“ s_coli”,代表大肠杆菌。使用“详细信息”超链接可视化箱形图。箱形图显示了与健康对照相比,CD患者中大肠杆菌更加丰富的趋势。
-
接下来,从“算法”下拉菜单中选择“ DESeq2”,然后单击“提交”。与edgeR相比,DESeq2是一种更为保守的算法,它可以将CD和对照组之间的2种物种识别为明显不同。这2个也都用edgeR识别。例如,大肠杆菌已经牵涉到IBD的发病机制,而副流感嗜血杆菌已显示在IBD中增加。
-
(可选)尝试使用不同的分类级别进行进一步的探索性分析。返回“分析视图”页面,并探索专门用于标记基因数据差异丰度分析的“metagenomeSeq”方法。
-
具有线性判别分析效果大小(LEfSe)的生物标志物发现。接下来,我们将使用LEfSe方法(框5)来鉴定CD组中稳定的生物标志物。返回“分析概述”页面,然后单击“ LEfSe”。上半部分包含分析参数,而下半部分包含两个选项卡。第一个选项卡是LEfSe结果的图形摘要,而第二个选项卡显示结果表。在参数面板中,将分类级别更改为“属”,将重要性阈值更改为“ 0.1”(经FDR调整或q值),然后单击“提交”。当使用以下临界值时,有11个分类单元被认为是重要的:q值<0.1和线性判别分析(LDA得分)> 2.0(图6)。
图6 LEfSe分析的图形简要展示
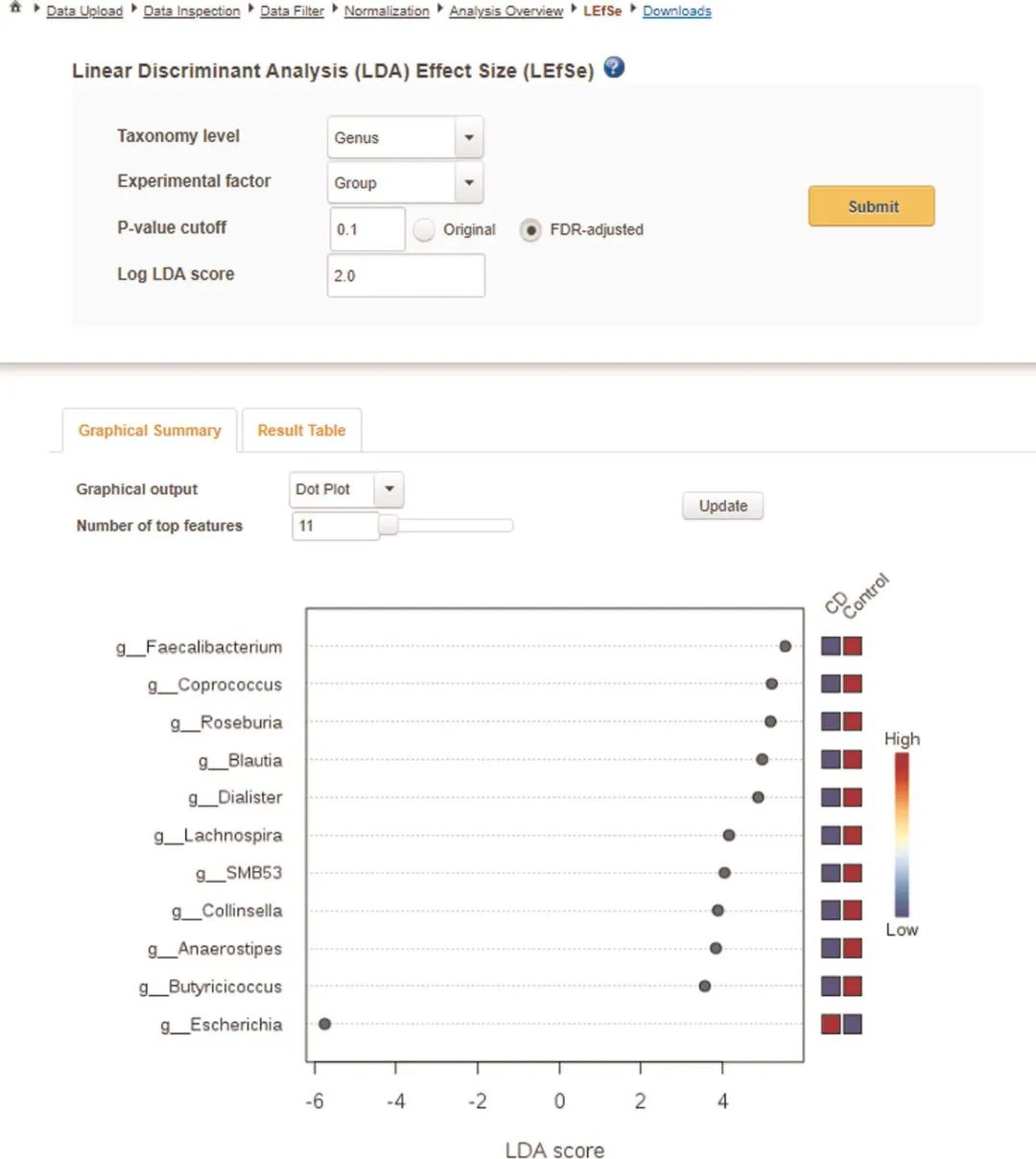
图6 LEfSe分析的图形。重要分类单元按其LDA分数(x轴)降序排列。绘图右侧的迷你热图指示每组中的分类单元是较高的(红色)还是较低的(蓝色)。
Fig. 6 | Graphical summary of LEfSe analysis. Significant taxa are ranked in decreasing order by their LDA scores (x axis). The mini heatmap to the right of the plot indicates whether the taxa are higher (red) or lower (blue) in each group.
-
默认情况下,图形输出显示的点图最多包含按其LDA分数排名的前15个要素。在“Number of top features”旁边的文本框中输入“11”,然后点击“更新”,以更改图形中包含的特征数量(图6)。从更新的图形摘要中,右侧的迷你热图指示了整个组中微生物特征的丰富程度。与健康对照相比,CD患者中10个属的类群减少,而大肠杆菌是CD患者中唯一增加的类群。对于多类数据集,图解的解释本质上是相同的。迷你热图将指示哪个类别中哪个分类单元最丰富。用户还可以选择查看条形图摘要(在“图形输出Graphical output”下拉菜单下),该摘要使用不同的颜色指示每种表型的最正相关的分类单元。
-
使用“随机森林”进行分类在“分析概述”页面上,单击“随机森林Random Forest”。随机森林(RF)算法是一种功能强大的机器学习方法,可以应用于微生物组数据以对重要特征进行分类和选择(框5)。默认情况下,RF模型是使用500棵树创建的。使用下拉菜单将其设置为“ 5000”,将“分类标准”设置为“属”,然后单击“提交”。在“分类效果”标签中,使用5,000棵树的实际(OOB)错误为0.14。由于算法的随机性,该值对于某些用户可能有所不同(图7)。该图显示了在属水平数据上训练的RF模型在预测CD或对照样品分类方面的性能。RF可以自然地处理多类数据集,并将为每个组计算OOB错误和分类性能。
图7 可视化“随机森林”结果
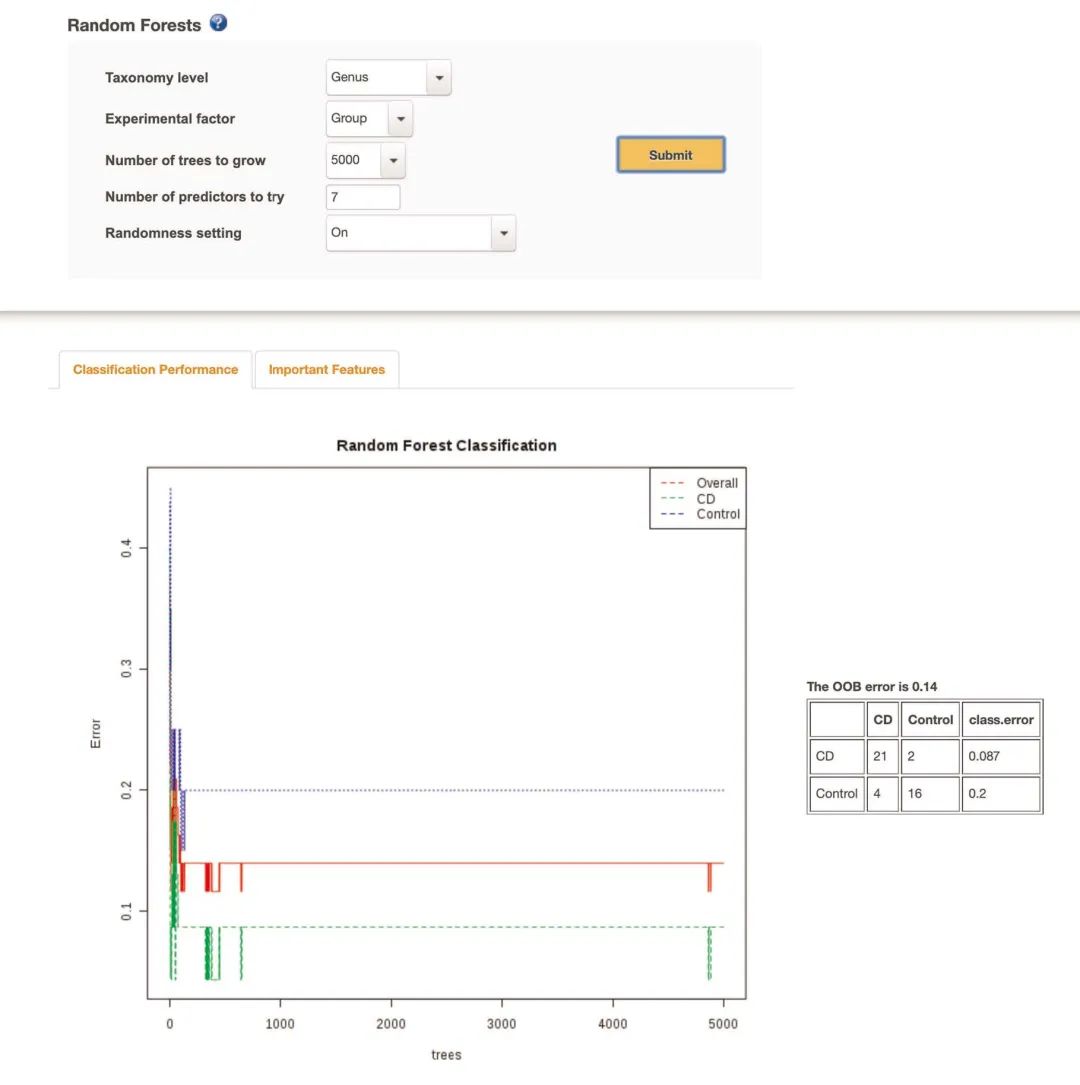
图7 | 可视化“随机森林”结果。“随机森林”分析结果的屏幕截图。右表显示了每个组的分类效果表格。用户可以点击“重要特征”标签查看那些对模型准确性有重大影响的标记物。
Fig. 7 | Visualization of the ‘Random Forests’ results. A screenshot of the ‘Random Forests’ analysis results. The classification performance for each group is shown in the table to the right. Users can click the ‘Important Features’ tab to view those features with large impact on the accuracy of the model.
-
使用RF识别重要功能。点击“重要功能Important Features”标签以查看图形结果。该图的布局与LEfSe图的布局相同(步骤26和27),不同之处在于按特征的平均下降精度(Mean Decrease Accuracy)对特征进行排名。对于多类数据集,迷你热图有助于可视化不同组之间的变化模式。使用LEfSe和RF,一贯认为Roseburia和无厌食杆菌(Anaerostipes caccae)显示出小儿CD与健康对照之间最重要的差异,其特征是CD患者的丰度降低。两种微生物都是丁酸盐的生产者,丁酸盐是一种具有已知抗炎作用的代谢物,其消耗与IBD有关。
-
分析报告的生成和结果下载。分析之后,点击顶部导航栏中的“下载Downloads”超链接。将显示“结果下载”页面,其中显示所有图形,结果表和“R命令历史记录”文件。单击“生成报告Generate Report”按钮,创建一个PDF报告,其中详细说明了所有已执行并嵌入结果的分析(图2)。点击“分析报告 Analysis Report”链接以下载报告。点击“ Download.zip”链接,下载包含分析会话中生成的所有结果的压缩文件。
阶段2:预测功能分析和基因丰度数据分析
Stage 2: Predictive functional profiling and analysis of gene abundance data
大约20分钟,具体取决于数据集的大小。
-
启动。返回MicrobiomeAnalyst主页,然后单击“标记数据分析(MDP)”以输入模块。
-
示例数据上传。从示例数据集中选择“ Aging Mouse Gut”,然后单击“ Submit”。重复步骤3-5,执行数据处理。
-
功能潜力的预测。通过重建未观察到的状态(PICRUSt)对群落进行系统发育研究是一种计算方法,可以从正确注释的OTU丰度表中预测基因丰度(更多详细信息,请参见框6)。点击“分析概述”页面中的“功能预测Functional prediction栏目下的“PICRUSt(Greengenes)”超链接。注意:PICRUSt的MicrobiomeAnalyst实现基于Greengenes参考OTU(2012年5月18日版本)。如果用户尚未将其数据注释到该数据库,则分析将失败。
-
在PICRUSt页面上,点击“预测功能潜力(Predict Functional Potential’)”按钮。此步骤将需要1〜2分钟,具体取决于当时的服务器负载。
-
完成后,页面上将显示所有样品中KO计数的箱形图。从“页面下载”菜单中,单击“ KO表(KO table)”和“元数据文件(Metadata File)”选项以下载这些文件。这些文件将用作SDP模块的输入。对于鸟枪法宏基因组学和宏转录组学数据,以下描述的过程同样有效。
-
返回MicrobiomeAnalyst主页,然后单击“宏基因组数据分析 Shotgun Data Profiling(SDP)”以进入该模块。
-
数据上传。要上传PICRUSt数据,请首先选择“上传基因丰度表(Upload a gene abundance table)”面板。在“基因ID类型(Gene ID type)”下拉菜单中,选择“KEGG Ortholog(KO)”。接下来,点击“Abundance file”旁边的“选择文件”按钮,找到“ functionalprof_picrust.csv”文件。按“打开”选择文件。接下来,点击“元数据文件Metadata file”旁边的“选择文件”按钮,然后找到“metadata.csv”文件。点击“提交”以上传所有数据。为了促进测试过程,我们还将这些数据包括在示例数据集中。要使用此功能,请点击页面底部的“示例数据集进行测试”。选择“ KO Mouse Dataset”,然后单击“提交”以上传数据。注:所需的格式是
.txt或.csv文件,其中基因在行中,样本在列中。可接受的基因标识符包括KO,酶委员会(EC)和直系同源簇(COG)。第一行必须包含示例名称,并以“#NAME”开头。可以使用用于MDP的相同元数据文件,第一列为样品名称,后跟元数据变量。点击“数据格式”页面以获取更多详细信息。 -
数据完整性检查。“数据完整性检查Data Inspection”页面总结了数据上传的结果。点击“继续Proceed”继续。
-
数据筛选。保留默认的“低计数过滤器low count filter”和“低差异过滤器low variance filter”设置,然后点击“提交Submit”。有关更多详细信息,请参阅步骤4。右上角将出现一条消息,指示剩余功能的数量。点击“已处理Procced”以继续。
-
数据标准化。保持“数据缩放scaling”设置为“总和缩放Cumulative sum scaling (CSS)”,然后点击“提交”。有关可用的标准化方法的详细信息,请参见框3。点击“继续”继续。
-
分析概述。“分析概述Analysis Overview”页面提供了一些功能分析,聚类分析,差异丰度分析和生物标志物分析的选项。步骤18-29涵盖了差异丰度分析和生物标记分析。在这里,我们将展示如何获得功能概述。点击“多样性概述Diversity overview”开始。
-
功能多样性分析。在“功能多样性分析Functional Diversity Profiling”页面上,用户可以通过将相关基因分为几个功能类别(包括KEGG metabolism,KEGG pathways,KEGG modules和COG Functional catalory)来查看其基因丰度数据潜在功能的图形摘要。页面顶部包含用于自定义绘图的参数,例如功能类别(Functional category)和配色方案(Color scheme)。默认视图是使用命中总数的KEGG代谢。从图中可以看出,在此功能级别上,年轻Yound,中年Mid和老年Old小鼠的细微差异。要探索不同的功能级别,请选择“KEGG途径”和“按类别大小归一化的总匹配数Total hits normalized by catalory size”。点击“提交”以更新图解。从新的堆叠区域图中,我们可以看到样品和条件之间不同途径的分布。
-
富集分析Enrichment analysis。我们可以进行富集分析,以统计学方式评估某些途径或模块是否与年龄因素显着相关。使用公认的全局测试算法计算富集,该算法是一种可靠的测试,可根据特定基因集(即KEGG途径)是否基于其丰度谱与表型转移显著相关。返回“分析概述”页面,然后单击“关联分析Association analysis”。将会出现一个弹出窗口。将实验系数设置为“年龄Age”,然后按“继续”。
-
使用KEGG全局代谢网络(global metabolic network)进行可视化。在“网络查看器Network Viewer”页面上,用户可以直观地浏览KEGG全局代谢网络中的富集途径(图8)。该页面包括三个部分:顶部工具栏,包含途径分析结果的左侧面板以及显示代谢网络的中心区域。要演示此页面的实用性,请点击“苯丙氨酸代谢Phenylalanine metabolism”旁边的复选框。现在,来自用户数据的匹配KO将在网络上突出显示为边缘,其颜色基于用户指定的突出显示颜色。
图8 KEGG整体代谢网络中富集通路的可视化
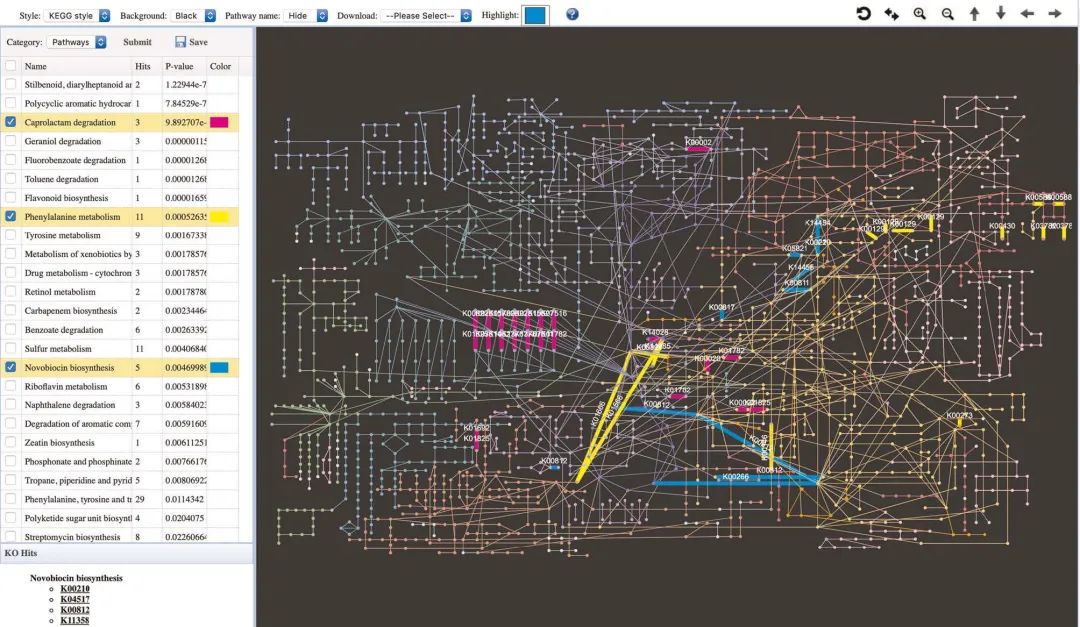
图8 | KEGG整体代谢网络中富集通路的可视化。KEGG整体代谢网络的屏幕截图。顶部工具栏包含用于网络自定义的所有选项,例如背景颜色,突出显示颜色以及是否显示路径名称。左侧面板包含浓富集分析的结果,面板底部提供了所有匹配KO的KEGG网站链接。通过在网络中以不同颜色突出显示标记路径。
Fig. 8 | Visualization of enriched pathways in the KEGG global metabolic network. A screenshot of the KEGG global metabolic network. The top toolbar contains all options for network customization, such as background color, highlight color, and whether to show pathway names. The left panel contains the results of the enrichment analysis, and the bottom of the panel provide links to the KEGG website for all matched KOs. Selected pathways are highlighted in different colors within the network.
-
网络探索。进一步探索富集分析的结果。用鼠标放大和缩小,以及向任何方向拖动网络。用户可以双击任何突出显示的边缘以查看相关的反应。页面的左下角列出了所选途径中所有匹配的KO。如果用户单击任何KO,它们将被直接带到KEGG网站上的相应页面。
-
网络定制。页面顶部的工具栏包含许多有用的选项,用户可以使用这些选项来自定义其网络。其中包括更改网络背景(Background: Black/White),显示或隐藏路径名称(Pathway name: Hide/Show)以及切换整体网络样式(Style)。调整这些设置以自定义网络。
-
进一步的网络定制。用户还可以用不同的颜色突出显示其指定的路径。例如,点击“突出显示(Hightlight)”旁边的彩色框。将出现一个调色板。直接点击显示感兴趣颜色的区域,然后按“选择Choose”以关闭对话框。接下来,点击左侧的“香叶醇降解Geraniol degradation”途径,突出显示所有匹配的边缘。
-
网络下载。进行网络探索之后,请点击“下载Download”旁边的下拉菜单,然后选择“PNG”图片。具有已创建网络的屏幕上将弹出“下载对话框”。右键单击PNG图像,然后将其保存为您的首选名称。或者,用户可以以SVG格式导出KEGG网络。
-
(可选)要进一步探索基因丰度数据(例如,差异丰度分析和生物标记分析),请执行步骤18-29。
阶段3:使用兼容的公共数据集进行可视化数据探索
Stage 3: Visual data exploration with a compatible public dataset
大约10分钟,具体取决于数据集的大小。
-
启动。返回MicrobiomeAnalyst主页,然后单击“带有公共数据的投影(PPD)”以进入该模块。
-
数据上传。PPD上传页面类似于MDP上传页面。点击“用于测试的示例数据集”以显示所有可用的示例数据集。选择“Arable soil”数据集。点击“提交”按钮上传数据。或者,单击“ ASV / OTU表”旁边的“选择文件”,然后找到“ soil_test_otu.txt”。单击“元数据文件”旁边的“选择文件”并找到“ soil_sample.txt”,然后单击“分类法”表旁边的“选择文件”并找到“ soil_test_taxa.txt”。将“分类标签”指定为“ Greengenes OTU ID”。点击“提交”以上传数据。
-
数据完整性检查。“数据完整性检查 Data Inspection”页面总结了数据上传的结果。点击“继续Proceed”继续。
-
数据选择 Data Selection。“数据选择”页面包含MicrobiomeAnalyst中所有可用的数据集,供用户与其数据共同投影。数据集由身体部位(用于人类样品),生物(来自其他哺乳动物的样品)和环境样品组成。由于示例数据来自可耕土壤,因此请点击“环境Environmental”标签以查看所有可用选项。选择“全球土壤‘Global soil”,然后点击“提交Submit”。注:用户数据和选定的公共数据集之间必须至少共享20%的分类单元。
-
交互式数据可视化。3D PCoA的外观应类似于图3。有关导航图的说明,请参阅步骤10–12。用户数据用圆圈表示,而公共数据则用正方形表示。使用鼠标旋转或放大或缩小图形。显然,来自用户数据的样本分为三个类,一个类接近于来自干燥土壤和表层土壤组的样本,而其他类则与所有参考数据相距甚远。
-
(可选)比较不同样本的分类单元数量。双击一个数据点(即一个样本)以查看其分类单元丰度的饼图摘要(步骤11和12)。请注意,所有生成的饼图都将出现在右侧的“查看历史记录”面板中。直观地比较这些饼图,以了解样品在不同生物分类水平下的不同情况。
-
分析下载。点击顶部导航栏中的“下载”链接以下载结果。
阶段4:分类单元列表的富集分析
Stage 4: Enrichment analysis of a list of taxa
时间约10分钟
-
启动。返回MicrobiomeAnalyst主页,然后单击“ Taxon Set Enrichment Analysis(TSEA)”以输入模块。
-
数据上传。所需格式是分类单元列表。使用“设备设置”部分中描述的示例分类清单列表文件(ibd_taxa.txt)。在您喜欢的文本编辑器(例如记事本)中打开“ ibd_taxa.txt”文件。选择所有分类单元名称,然后将内容复制并粘贴到MicrobiomeAnalyst的文本区域中。将“输入类型Input type”保留为“混合级别的分类名称Mixed-level Taxon Names”。点击“提交Submit”按钮。注:用户必须将其分类单元列表作为分类单元名称或ID的单列上载,并正确指定输入类型才能继续。
-
名称映射Name mapping。下一页显示“ Taxonomic Name / ID Mapping”功能的结果。该页面的目的是将用户数据中的分类单元名称与MicrobiomeAnalyst的基础分类单元集库进行匹配。没有命中的分类单元名称将以黄色突出显示,并将被排除在进一步分析之外。点击页面底部的“提交”按钮继续。
-
“Taxon Set Library”页面显示了所有可用于富集分析的分类单元。分类单元集分为三个级别:“混合级别”(包括门到种),“物种级别”和“菌株级别”。在此例中,分类单元是属名和种名的混合。在“混合级别的分类单元集”标题下,单击“宿主内在的分类单元集Host-intrinsic taxon sets”,然后单击“提交”继续进行下一步。
-
网络探索。TSEA结果显示为富集网络(图9)。在网络中,每个节点代表一个分类单元集,其颜色对应于P值,其大小对应于可比对次数。如果共享分类单元的数量> 20%,则连接两个节点。在网络的基础上,“小儿克罗恩氏病Pediatric Crohn’s Disease”获得的出现次数最多,并且与其他分类群(例如“ 1型糖尿病Type 1 Diabetes’”,“结直肠癌变Colorectal carcinogenesis”,“克罗恩氏病Crohn’s Disease”和“厌食症(减少)‘Anorexic (decrease)’”高度相关 。拖动节点周围或使用鼠标滚动来放大或缩小。点击任一分类单元集,以在右侧面板的“Taxon Set View”中查看其详细信息。所有匹配的分类单元将以红色突出显示。指向相应出版物的链接以指向PubMed以及出版物中证据收集地的超链接形式提供。
图9 TSEA结果
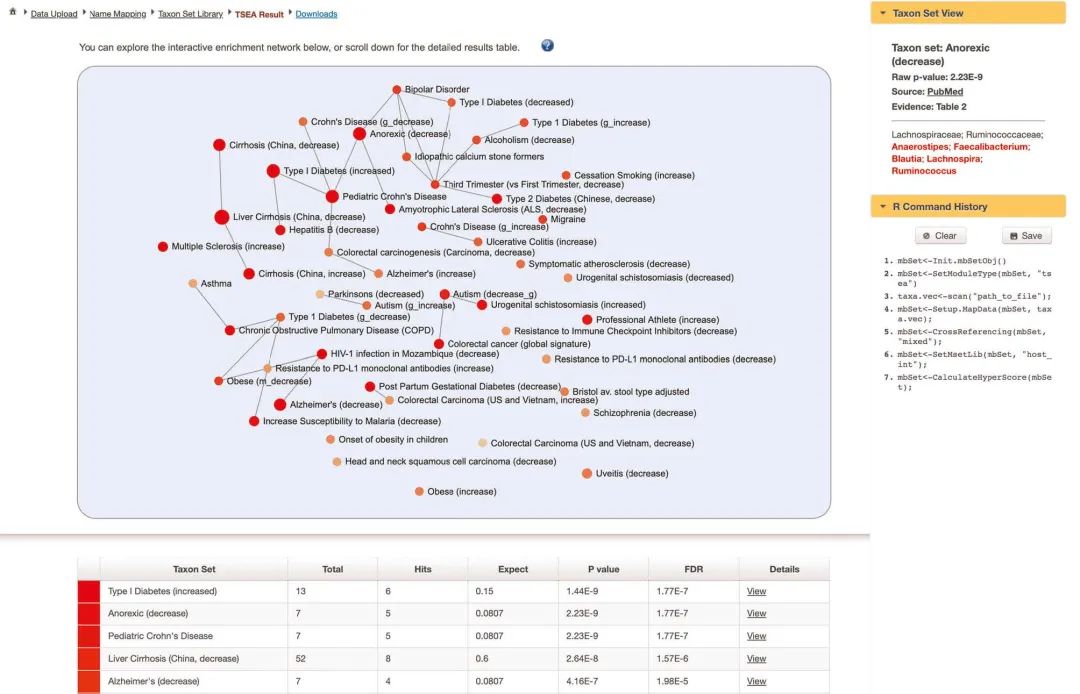
图9 | TSEA结果。页面顶部是一个可缩放的网络。用户可以单击任意节点,以通过右侧的“ Taxon Set View”选项来查看有关基础分类单元集的更多详细信息。带有详细统计信息的结果表显示在页面底部。
Fig. 9 | TSEA results. At the top of the page is an enrichment network. Users can click any node to view more details about the underlying taxon set via the ‘Taxon Set View’ option on the right. The result table with detailed statistical information is shown at the bottom of the page.
-
探索TSEA结果表。向下滚动页面以查看结果表。十个分类单元集的FDR调整后的P值<0.05。“厌食症Anorexic”是最丰富的分类单元集之一。这并不意外,因为营养不良是小儿IBD的常见并发症,可能源于厌食症。花一些时间探索TSEA的其他结果。
-
下载结果。点击顶部导航栏中的“下载”链接,进入“结果下载”页面。生成相应的分析报告并下载结果。点击“退出”退出会话。
框3 数据筛选和标准化
Box 3 | Data filtering and normalization

此框描述了MicrobiomeAnalyst中可用于数据过滤和标准化的不同方法。微生物组数据会受到从样品制备到测序的各种系统变异的影响。过滤和标准化的目的是消除或减少这种系统的可变性。下面将进一步讨论最常用方法的优缺点。但是,方法的选择取决于要执行的分析的类型。
数据过滤
数据过滤的目的是消除低质量和/噪音,以改善下游统计分析。MicrobiomeAnalyst提供了三种数据过滤程序(i)最小数据过滤(适用于所有分析),可删除包含全零或仅出现在一个样本中的特征;(ii)低频序列过滤,删除可能由于测序错误或污染而存在的特征 ; (iii)低方差过滤,它消除了不太可能与研究条件相关的特征。最后两个选项不用alpha多样性,但强烈建议用于差异比较分析。
数据抽平
抽平通常用于解决不均匀的文库大小。此方法通过随机进行二次采样而不替换不认为有缺陷的最小文库的大小。由于有用信息的潜在丢失而受到批评。但是,该方法已被证明对于组之间非常小的(<1,000个读长/样本)或非常不均匀的库大小(差异大于> 10倍)有用,对于比较生态群落(β多样性)也很重要。
数据缩放
缩放涉及将特征数量乘以样本比例因子以解决不均匀的测序深度,将原始读长频数转换为相对丰度。最常用的方法是总和缩放(total sum scaling, TSS),其中数据除以每个样本中读长的总数。该方法受到批评,因为读长数量的总数可能受少数几个最丰富的特征所支配,这会导致相对丰度不够准确。此外,TSS并未考虑跨测量值的特征方差的异方差性。已经提出了其他标准化方法,例如:高分位数(upper quantile, UQ)和累积总和缩放(cumulative sum scaling, CSS),以解决此类问题。特别是在进行差异分析时,建议使用CSS来控制具有较大组大小的数据中的FDR。但是,在进行群落级别的比较(例如估算β多样性)时,建议使用TSS,因为它可以最准确地捕获原始群落的组成,而UQ和CSS会使群落变形。
数据转换
数据转换的目的是稳定数据的方差。由于微生物组数据性质,通常建议使用中心对数比(centered log ratio,CLR)。此外,其变体,相对对数表达(relative log expression,RLE)和修整均(mean,M)值(TMM)在识别差异特征方面一直表现出较高的性能。
框4 Alpha和beta多样性
Box 4 | Alpha and beta diversity

此框描述了MicrobiomeAnalyst中可用于群落分析的α和β多样性分析。Alpha多样性是样本内多样性的一种度量,而β多样性是样本间多样性的一种度量。可以将Alpha多样性视为单个样本多样性的汇总统计,而可以将β多样性估计值视为成对样本之间的相异性结果。对于后者,这些措施允许通过聚类或降维技术进行进一步分析。可以使用各种统计检验来评估差异是否显着。更多细节请往下看。
Alpha多样性
Alpha多样性概括了样本中物种的丰富度(物种总数)和/或均匀性(物种间的丰度分布)。MicrobiomeAnalyst当前支持六种alpha多样性度量,每种评估群落的不同方面。“Observed”可计算每个样本的特征总数,而“ACE”和“ Chao1”可通过计算由于丰度低而未被检测到的特征来估算分类单元丰富度。“Shannon”和 “Simpson”考虑了物种的丰富性和均匀性,对均匀性的重视程度各不相同。最后,“Fisher”将群落的丰度结构转化为对数。
Beta多样性
Beta多样性评估样本之间群落组成的差异。可以将所得的β多样性估计值合并到距离矩阵中,并用于进行排序。彼此接近的样品在其微生物群落特征方面更为相似。
MicrobiomeAnalyst支持五种最常用的β多样性指标。“Jaccard距离”仅使用特征的存在与否来计算微生物成分的差异;“Bray-Curtis差异”使用丰度数据并计算特征丰度的差异;“Jensen-Shannon divergence’”评估两种概率分布之间的距离,这些概率分布说明了微生物特征的存在与丰富度;“Unweighted UniFrac”和“weighted UniFrac”使用特征之间的系统发生距离-前者仅基于系统发生距离,而后者则根据特征的相对丰富度进一步加权。
可以使用PCoA或非度量多维标度(NMDS)可视化Beta多样性度量。两种方法都以距离矩阵为输入;PCoA使样本之间的线性相关性最大化,而NMDS使样本之间的秩相关性最大化。如果样本之间的距离太近可以PCoA进行线性变换更适合。如果用户希望突出显示其数据中的梯度结构,则建议使用NMDS。NMDS是迭代的,并且对于同一数据集可能返回不同的结果。此外,MicrobiomeAnalyst可以计算NMDS图的应力值(也就是我们常说的stress),这是拟合优度的度量。通常,> 0.2的值表示拟合效果较差,而<0.1的值表示拟合效果较好。使用PERMANOVA,组相似性分析(ANOSIM)或组分散均匀性(PERMDISP)评估组之间的协调措施的统计意义。这些测试评估了各组之间微生物组组成的总体差异。PERMANOVA测试所有组之间距离是否相等。它使用同一组样本之间的距离(或相异度),并将它们与组之间的距离进行比较。该方法对多元离散敏感。因此,还应使用PERMDISP评估样品之间的分散度(或变化)是否与组之间的分散度不同。ANOSIM使用所有成对样本距离的等级来测试组内距离是否大于或等于组间距离。
框5 相关、差异和分类
Box 5 | Correlation, comparison and classification

此框描述了MicrobiomeAnalyst中提供的相关(SparCC),差异比较(LEfSe)和分类(RF)分析方法。
相关性
相关网络的目的是确定微生物之间潜在的相互作用,这些相互作用可以表示相互关系,共生关系,寄生关系甚至竞争关系。揭示这种相互作用可能对微生物群落的健康具有重要意义,并最终促进对微生物组功能的理解。存在几种用于计算相关性网络的简单方法,例如Pearson相关性(用于确定两个分类单元之间是否存在线性关系)以及Spearman和Kendall的等级相关性(用于测量对之间的等级关系)。但是,这些简单的方法通常无法解决微生物组数据的组成性质,并且由于识别出虚假的相关性而可能不可靠。因此,已经引入了更加稳健的方法,例如SparCC和稀疏逆协方差估计以进行生态联系和统计推断(SPIEC-EASI)6,这两种方法都充分假设了稀疏相关网络。SparCC使用对数比率转换并执行多次迭代,以识别与背景相关性离群的分类单元对。SPIEC-EASI使用图形网络模型推断整个相关网络。两种方法都需要大量计算,之前最近引入了名为FastSpar可以将SparCC算法的高效实现。而我们的 MicrobiomeAnalyst封装了FastSpar以及Pearson,Spearman和Kendall的相关分析方法。来尝试不用换工具,即可在R中飞驰的感觉。
LEfSe 分析
LEfSe是一种非参数统计方法,旨在识别各组之间存在显着差异的微生物分类群。首先,使用Kruskal-Wallis检验来识别其相对丰度在组之间显着不同的分类单元。然后将LDA应用于达到显着性阈值的分类单元,以估计其分类影响大小。此方法根据其LDA分数输出分类单元的排序列表。P < 0.05的显著性水平和LDA得分2通常用于确定最能表征每种表型的分类单元。最初的LEfSe实现可在Huttenhower Galaxy(https://huttenhower.sph.harvard.edu/galaxy)上获得,在执行LEfSe时会考虑整个分类单元(所有分类等级)。相比之下,MicrobiomeAnalyst实现仅在用户指定的分类级别上执行LEfSe。另外,最初的LEfSe实现在确定重要分类单元时使用原始P值。MicrobiomeAnalyst实现为用户提供了使用原始值或FDR调整后的P值临界值的选项。
随机森林
随机森林(Random Forest, RF)是一种有监督的机器学习算法,已应用于微生物组数据进行分类以及识别对分类具有重要作用的微生物分类群。RF非常适合用于大型和大噪音的数据,例如微生物组的数据,因为它能够识别非线性关系,处理可变的相互作用并且可以处理过度拟合。RF通过使用随机选择的训练数据子集构建多个决策树来工作。每棵树是通过在每个节点上随机选择一小组要分割的特征而形成的。通过所有树的多数投票来实现分类预测。为了评估分类准确性,在树构建过程中将1/3个样本去除,随后使用模型对这些样本进行分类,以计算袋外或OOB错误率。
框6 功能预测
Box 6 | Functional prediction

此框描述了MicrobiomeAnalyst中可用于预测功能的方法。
尽管它们可以通过分类信息得到功能信息,但标记基因数据并未直接提供任何功能信息。但是,从16S rRNA测序数据推断潜在功能还是非常有吸引力。MicrobiomeAnalyst提供两种建立完善的预测性功能预测方法:PICRUSt和Tax4Fun。PICRUSt是第一个普及从16S rRNA数据推断微生物组功能的方法的工具。它利用了系统发育相关生物更有可能具有相似基因含量的思路。从16S rRNA数据中,PICRUSt算法搜索具有注释基因组的最密切相关的生物,并假设其功能信息也存在于该数据中。另一方面,Tax4Fun是一个R程序包,它结合了来自KEGG原核生物的预计算的功能配置文件和标准化的分类学丰度。要使用Tax4Fun,必须使用SILVA参考数据库注释输入的16S rRNA测序数据,而对于PICRUSt,必须使用Greengenes数据库。两种方法都依赖于可用的基因组注释进行推论,并且适用于来自的环境(如人类肠道)中的微生物的预测功能分析。
故障排除Troubleshooting
故障排除建议可在表2中找到。
表2 常见问题

| 步骤 | 问题 | 可能原因 | 解决方案 |
|---|---|---|---|
| 1 | 主页显示不正常 | 浏览器禁用了JavaScript | 搜索你使用浏览器打开JavaScript的方法。如谷歌Chrome,点击右上角的3个点菜单中点”设置”,滑至底部点”高级”,在隐私设置和安全性栏目,点网站设置,点JavaScript,再点允许。 |
| 2 | 数据上传失败 | 没选中,或格式错误 | 根据提示消息显示可能原因,(i)非制表符分隔;(ii)错误选择分类标签;(iii)使用非分号分隔分类单元;(iv)格式不支持 |
| 3 | 数据完整性检查失败 | 样品名与丰度表不匹配;分类单元在有重名 | 确保样品在所有上传文件中一致;确保特征表与分类特征注释编号一致 |
| 60 | 上传的分类单元不匹配;一段时间后服务器响应 | 尽管存在大量的分类学数据库,但不可能包括所有微生物;系统默认45分钟会超时退出 | 我们会增加分类单元扩展微生物组的覆盖度;刷新并重新上传数据,我们正在使用帐户管理系统,使用户可以保存并恢复分析 |
时间Timing
第1步至第30步,阶段1,对16S丰度数据进行全面分析:约30分钟,具体取决于数据集的大小
步骤31-49,阶段2,预测功能分析和基因丰度数据分析:〜20分钟,具体取决于数据集的大小
步骤50-56,阶段3,使用兼容的公共数据集进行可视数据探索:〜10分钟,具体取决于数据集的大小
步骤57-63,阶段4,分类单元列表的富集分析:〜10分钟
预期结果Anticipated Results
该方法使用户能够对其微生物组数据进行全面分析。提供了三个示例数据集:每个分别用于儿童IBD样本,老年小鼠样本和耕地土壤样本。在分析过程中产生的主要图形输出如图3–9。用户不仅能够描述其微生物群落并识别重要特征,还可以通过富集分析和基于代谢网络的可视化获得功能见解。PPD和TSEA模块还允许用户通过将其数据与兼容的公共数据集或已知的微生物标签进行比较来进行潜在分析或新颖见解,从而进行元分析。
报告摘要Reporting Summary
链接到本文的《自然研究报告摘要》中提供了有关研究设计的更多信息。
数据可用Data availability
方法中使用的所有示例数据集均作为示例数据集集成在其各自的模块中,也可以从MicrobiomeAnalyst的“资源”页面(https://www.microbiomeanalyst.ca/MicrobiomeAnalyst/docs/Resources.xhtml)下载。对它们的使用没有限制。
译者:文涛 南京农业大学
责编:刘永鑫 中科院遗传发育所
Reference
-
Jasmine Chong, Peng Liu, Guangyan Zhou & Jianguo Xia. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nature Protocols. 2020, 15: 799-821. doi:10.1038/s41596-019-0264-1
-
MicrobiomeAnalyst:可视化微生物组网页工具
-
NAR:MicrobiomeAnalyst——统计、可视化微生物组数据
-
Nature子刊:使用MicrobiomeAnalyst分析微生物组数据 https://www.mr-gut.cn/papers/read/1067040285
-
MicrobiomeAnalyst 网页交互式分析微生物组数据神器:https://mp.weixin.qq.com/s/mcGsGgF5aJ4M4FQ-Vw6FNw
这篇关于Nature子刊:教你零基础开展微生物组数据分析和可视化的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!







