本文主要是介绍破布叶(Microcos paniculata)单倍型染色体级别基因组-文献精读22,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
Haplotype-resolved chromosomal-level genome assembly of Buzhaye (Microcos paniculata)
破布叶、布渣叶(Microcos paniculata)单倍型解析染色体级别基因组组装

摘要
布渣叶(Microcos paniculata)是一种传统上用作民间药物和制作草药茶的灌木。之前对该物种的研究主要集中在其化学成分和药用价值上。然而,缺乏参考基因组限制了对该物种活性化合物分子机制的研究。在此,我们基于PacBio HiFi和Hi-C数据组装了M. paniculata的单倍型解析染色体级别基因组。组装包含两个单倍体基因组,大小分别为399.43 Mb和393.10 Mb,Contig N50长度分别为43.44 Mb和30.17 Mb。约99.93%的组装序列可以锚定到18条伪染色体。此外,共鉴定出482 Mb的重复序列,占基因组的60.76%。共鉴定出49,439个蛋白编码基因,其中48,979个(99%)得到了功能注释。该单倍型解析染色体级别的组装和注释将成为研究该物种活性化合物的生物合成和遗传基础的重要资源,并推动锦葵目进化基因组学研究的发展。
背景与概要
布渣叶(Microcos paniculata Linnaeus)(图1a),在中文中称为布渣叶,是一种在传统中药和凉茶中常用的灌木1,包括王老吉、霍七正2和加多宝,年需求量约为250吨(布渣叶后市浅析--A03市场周刊·行情分析--2008-04-28--中国医药报)。布渣叶的叶子也常用于治疗食积、湿热黄疸和发热的民族药物中3。迄今为止,许多研究广泛调查了该物种的植物化学成分和药理特性,揭示了布渣叶提取物中存在的生物活性次级代谢产物,如黄酮类、生物碱、三萜类和有机酸1,4。然而,由于缺乏高质量的参考基因组,布渣叶中次级代谢产物生物合成的分子基础和进化很少被报道5。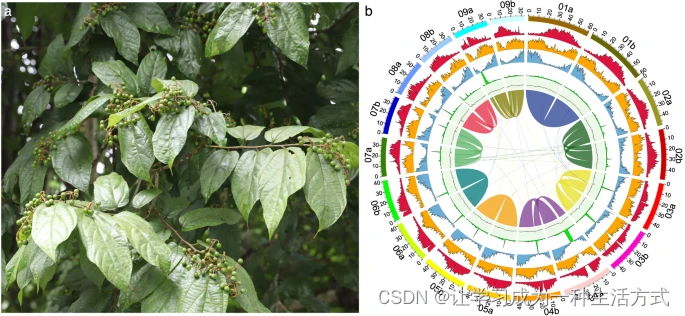
形态特征 (a) 和布渣叶 (M. paniculata) 基因组组装和注释的景观 (b)。从外到内的轨道依次为:伪染色体、I类转座子密度、II类转座子密度、蛋白质编码基因密度、串联重复比例、GC含量和共线性区块。 在本研究中,我们使用106×短读段(42 Gb)、35×HiFi读段(14 Gb)、75×Hi-C读段(30 Gb)和50×iso-seq读段(20 Gb)对布渣叶基因组进行了组装。最终的组装(约792 Mb)由两个完整的单倍型组成,单倍型A(399.43 Mb)和单倍型B(393.10 Mb),contig N50长度分别为43.44 Mb和30.17 Mb(表1)。约99.93%的组装序列锚定到18(2n)条伪染色体上(图1b)。叶绿体和线粒体基因组分别为159,456 bp和380,905 bp。总共鉴定出1,080,648条重复序列,长度约为482 Mb,占组装基因组的60.76%。在鉴定的重复序列中,长末端重复序列(LTRs)占最大比例,数量为394,112,累积长度为321,160,287 bp,占布渣叶基因组组装的40.52%(表2)。基因组包含65,874个基因,包括49,439个蛋白质编码基因和16,435个非编码基因(表3)。共注释了48,979个基因,占鉴定出的蛋白质编码基因的99%(表4)。其中,44,971个基因通过三种方法共同注释(图2)。特别是,有639个基因被注释为与黄酮类、生物碱和三萜类的生物合成或代谢相关(表S1)。布渣叶的高质量参考基因组和注释将成为提高我们对锦葵目进化关系理解的重要资源,用于研究植物化学化合物的分子基础和生物合成机制,并进一步研究和开发布渣叶。
Summary of M. paniculata genome assembly.
| Parameter | Genome | Haplotype A | Haplotype B |
|---|---|---|---|
| Genome size | 792,535,851 bp | 399,432,223 bp | 393,103,628 bp |
| GC content | 35.74% | 35.73% | 35.75% |
| Contig number | 37 | 18 | 19 |
| Contig N10 | 49,527,071 bp | 55,167,130 bp | 49,527,071 bp |
| Contig N50 | 41,049,410 bp | 43,438,762 bp | 30,170,985 bp |
| Contig N90 | 12,203,702 bp | 13,880,047 bp | 12,203,702 bp |
| Scaffold number | 20 | 11 | 9 |
| Scaffold N10 | 60,658,723 bp | 60,706,172 bp | 60,658,723 bp |
| Scaffold N50 | 45,573,016 bp | 47,575,556 bp | 45,573,016 bp |
| Scaffold N90 | 35,541,173 bp | 35,541,173 bp | 36,361,311 bp |
| Gap number | 17 | 7 | 10 |
Summary of repeat elements.
| Type | Number | Length (bp) | Percent (%) | Mean length (bp) |
|---|---|---|---|---|
| LTRs | 394,112 | 321,160,287 | 40.52 | 815 |
| LINE | 5,466 | 3,375,940 | 0.43 | 618 |
| Helitron | 154,911 | 42,417,336 | 5.35 | 274 |
| TIR | 188,121 | 59,996,054 | 7.57 | 319 |
| Unclassified | 132,568 | 45,978,909 | 5.8 | 347 |
| Simple repeats | 172,726 | 7,029,166 | 0.89 | 41 |
| Low complexity | 32,712 | 1,583,109 | 0.2 | 48 |
| Polinton | 32 | 5,983 | 0 | 187 |
| Total | 1,080,648 | 481,546,784 | 60.76 | 446 |
Summary of M. paniculata genome annotations.
| Feature | Total | Haplotype A | Haplotype B |
|---|---|---|---|
| gene | 65,874 | 37,351 | 28,523 |
| transcript | 76,776 | 42,840 | 33,936 |
| CDS | 60,341 | 30,283 | 30,058 |
| exon | 363,716 | 187,057 | 176,659 |
| intron | 286,940 | 144,217 | 142,723 |
| mRNA | 49,439 | 24,794 | 24,645 |
| rRNA | 14,488 | 11,547 | 2,941 |
| tRNA | 911 | 478 | 433 |
| other ncRNA | 1,036 | 532 | 504 |
Functional annotation of protein-coding genes in M. paniculata.
| Program | Database | Number | Percent (%) |
|---|---|---|---|
| eggNOG-mapper | GO | 22,963 | 46.45 |
| KEGG_KO | 22,373 | 45.25 | |
| EC | 10,045 | 20.32 | |
| KEGG_Pathway | 14,133 | 28.59 | |
| eggNOG | 44,508 | 90.03 | |
| COG | 47,855 | 96.80 | |
| DIAMOND | Swiss-Prot | 36,400 | 73.63 |
| TrEMBL | 48,572 | 98.25 | |
| NR | 48,206 | 97.51 | |
| TAIR10 | 43,580 | 88.15 | |
| InterProScan | CDD | 16,560 | 33.50 |
| Interpro | 42,031 | 85.02 | |
| Gene3D | 34,296 | 69.37 | |
| PRINTS | 7,479 | 15.13 | |
| Pfam | 39,734 | 80.37 | |
| SMART | 15,177 | 30.70 |
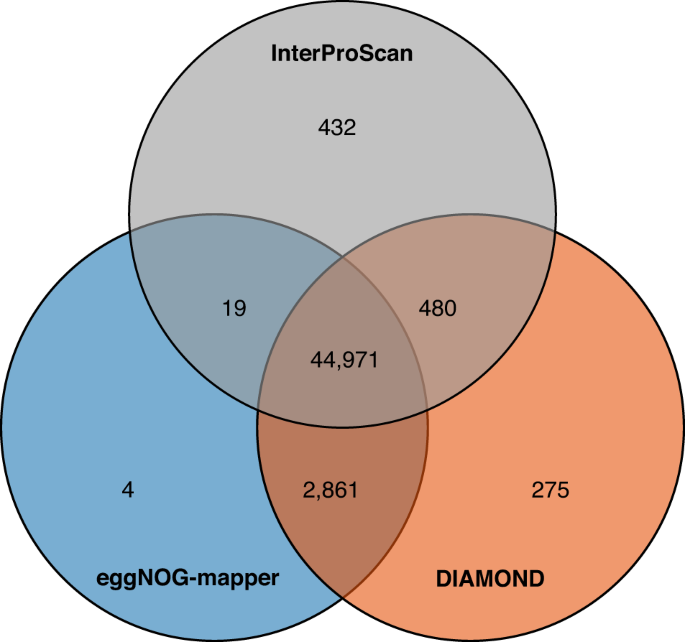
文氏图显示了使用三种策略在布渣叶中唯一和共享的功能注释蛋白质编码基因。
方法
样本采集与基因组测序
布渣叶(M. paniculata)的样本在中国云南省勐腊县中国科学院西双版纳热带植物园(XTBG)采集。使用改良的CTAB方法提取基因组DNA6。使用NanoDrop One分光光度计(NanoDrop Technologies,Wilmington,DE,USA)和Qubit 3.0荧光计(Life Technologies,Carlsbad,CA,USA)评估DNA质量。全基因组测序、Pacbio测序、Hi-C(高通量染色体构象捕获)测序和全长同源异构体测序(iso-seq)在武汉贝纳基科技有限公司(武汉,中国)进行。
对于全基因组测序,使用声波仪(Covaris,Brighton,UK)将1 μg基因组DNA超声处理至200-400 bp左右的大小范围。按照制造商说明构建短读段文库,然后使用PE(成对末端)150模式在DNBSEQ-T7平台(BGI lnc.,深圳,中国)上进行测序。
对于长读段测序,使用Megaruptor 3剪切套件(Diagenode SA.,Seraing,Belgium)剪切基因组DNA。使用AMPure PB磁珠选择套件(Pacbio,Menlo Park,CA,USA)选择性去除小于5 kb的DNA片段。使用SMRTbell®制备套件3.0(Pacbio,Menlo Park,CA,USA)制备文库,然后在Revio系统(Pacbio,Menlo Park,CA,USA)上进行测序。使用CCS工作流7.0.07参数(--streamed --log-level INFO --stderr-json-log --kestrel-files-layout--min-rq 0.9 --non-hifi-prefix fail --knrt-ada --pbdc-model)将原始测序数据转换为HiFi(高保真)读段。
对于Hi-C测序,将幼芽的叶片材料固定在2%甲醛溶液中,并按照已发表的协议8生成Hi-C文库。简而言之,交联材料用400单位MboI消化,并标记为生物素-14-dCTP,然后进行交联片段的平末端连接。重新连接后,逆向交联和纯化,通过声波处理将染色质DNA剪切至200-600 bp大小。然后使用链霉亲和素磁珠富集生物素标记的Hi-C片段。加入A尾和适配子后,对Hi-C文库进行PCR扩增(12-14周期),然后在DNBSEQ-T7平台(BGI lnc.,深圳,中国)上使用PE150模式进行测序。
全长同源异构体测序(iso-seq)用于获取高质量的转录组数据。使用R6827植物RNA提取试剂盒(Omega Bio-Tek,Norcross,GA,USA)按照制造商说明从布渣叶的叶、花和茎中提取RNA。使用牛津纳米孔(Oxford Nanopore Technologies,Oxford,UK)公司提供的cDNA-PCR测序试剂盒SQK-PCS109制备全长cDNA文库。然后在PromethION测序仪(Oxford Nanopore Technologies,Oxford,UK)上进行测序。
基因组组装
将PacBio HiFi读段和Hi-C短读段作为输入结合到Hifiasm v0.19.5-r5929中,使用默认参数生成单倍型解析的contig以进行后续分析。使用Juicer v1.5.610将Hi-C读段映射到组装的单倍型contig上,然后使用3D-DNA v18092211管道(参数为--early-exit -m haploid -r 0)进行Hi-C辅助的初始染色体组装。然后使用Juicebox v1.11.0812手动调整染色体边界并纠正错误连接和切换错误。这个过程生成了染色体级别的框架和未锚定的contig序列。
使用LR_Gapcloser v1.1.113基于HiFi读段填补染色体组装中的空隙(参数为-s p -r 2 -g 500 -v 500 -a 0.25)。然后将HiFi读段重新映射到染色体框架。将定位在端粒重复序列(TTTAGGG)周围的映射读段提取并使用Hifiasm v0.19.5-r592的默认参数组装成contig。将得到的contig重新对齐到染色体框架上以扩展染色体端部的端粒序列,共获得28个端粒序列(图3a)。此外,使用GetOrganelle v1.7.515组装叶绿体和线粒体基因组。
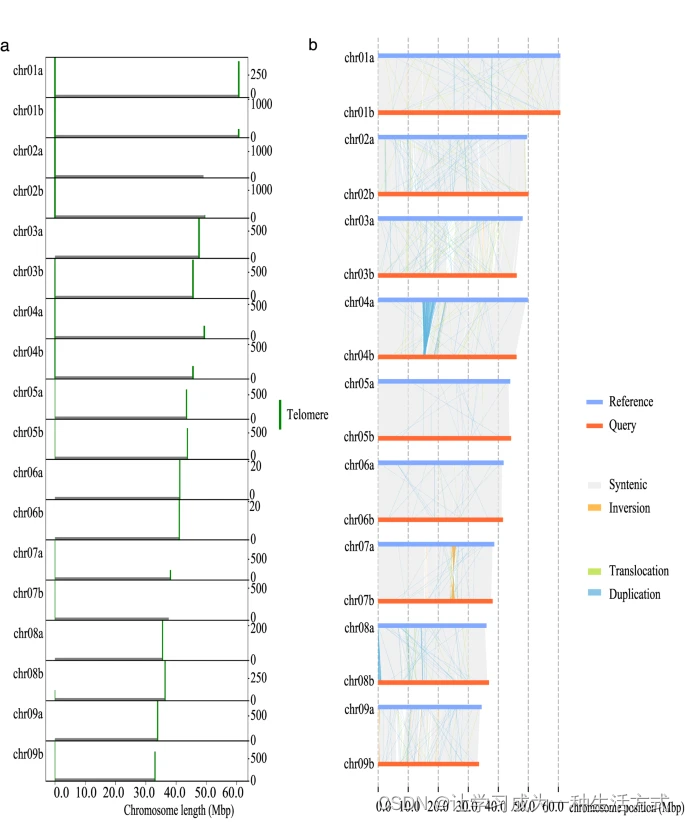
端粒分布 (a) 和单倍型A与单倍型B之间基因组结构的比较 (b)。 使用Nextpolish2 v0.1.016基于HiFi读段和短读段对上述组装进行了打磨,使用默认参数。通过Redundans v0.13c17管道(参数为-identity 0.98 -overlap 0.8)去除冗余单倍型和rDNA片段,并手动整理。最终获得了高质量的布渣叶单倍型解析基因组组装。
重复序列注释
使用EDTA(Extensive de novo TE Annotator)程序 v1.9.918(参数为--sensitive 1 --anno 1)进行转座子(TE)的新发现,生成TE库。使用RepeatMasker v4.0.719识别重复元素(参数为-no_is -xsmall)。
蛋白质编码基因和非编码RNA的注释 使用来自Theobroma cacao20、Durio zibethinus21、Corchorus capsularis22、Gossypium raimondii23、Heritiera littoralis24、Dipterocarpus turbinatus25、Aquilaria sinensis26、Arabidopsis thaliana27、Carica papaya28、Vitis vinifera29和Bombax ceiba30的314,962个公开的非冗余蛋白质序列作为同源蛋白质证据进行基因注释。使用Minimap2 v2.2431(参数为-a -x splice --end-seed-pen = 60 --G 200k)将iso-seq数据映射到基因组,然后使用StringTie v1.3.532(参数为-L -t -f 0.05)进行组装,所得序列用作转录证据。
使用PASA(Program to Assemble Spliced Alignments)v2.4.133根据转录证据注释基因组结构,使用默认参数。然后,通过与同源蛋白质证据对齐(使用BLAT34 -prot)并移除查询或目标覆盖率<95%的命中,鉴定出全长基因序列。使用AUGUSTUS v3.4.035通过全长基因集进行五轮训练和优化,使用默认参数。
使用MAKER2 v2.31.936管道基于ab initio预测、转录证据和同源蛋白质证据进行注释。简要说明:(1)使用RepeatMasker v4.0.719对基因组中的重复序列进行屏蔽;(2)使用AUGUSTUS v3.4.035基于基因组序列进行ab initio预测;(3)使用BLASTN将转录证据对齐到重复屏蔽基因组,使用BLASTX将同源蛋白质证据对齐到基因组。使用Exonerate v2.2.037将BLAST命中重新对齐到基因组;(4)最终,使用MAKER2根据上述对齐生成的提示整合预测的基因模型。
使用EvidenceModeler(EVM)v1.1.138进一步合并从PASA v2.4.1和MAKER2 v2.31.9获得的注释结果,生成共识注释。使用TEsorter v1.4.139识别基因组上的TE蛋白质结构域(参数为-genome -db rexdb -cov 30 -eval 1e-5 -prob 0.9),并在EVM过程中屏蔽这些结构域。通过整合UTR序列和可变剪接,使用PASA v2.4.1(默认参数)对EVM的结果进行优化。排除过短(<50个氨基酸)、缺乏起始或终止密码子、包含内部终止密码子或有模糊碱基的注释。然后合并所有注释,并移除冗余注释。
此外,对于非编码RNA(ncRNA)注释,使用tRNAScan-SE v1.3.140识别转移RNA(tRNA),使用Barrnap v0.9(GitHub - tseemann/barrnap: :microscope: Bacterial ribosomal RNA predictor)识别核糖体RNA(rRNA)。为了确保准确性,排除部分rRNA注释。此外,使用RfamScan v14.241识别其他ncRNA。
我们使用三种策略预测蛋白质编码基因的功能:(1)使用eggNOG-mapper v2.0.042(参数为--target_taxa Viridiplantae -m diamond)在eggNOG数据库中搜索同源基因,从而进行Gene Ontology(GO)和京都基因与基因组百科全书(KEGG)注释;(2)使用DIAMOND v0.9.2443(参数为--evalue 1e-5 --max-target-seqs 5)将蛋白质编码基因与Swiss-Prot、TrEMBL、NR(NCBI中的非冗余蛋白质)和TAIR10蛋白质数据库进行对齐;(3)使用InterProScan v5.27-66.044通过搜索多个公开数据库(如PRINTS、Pfam、SMART、PANTHER和InterPro数据库的CDD)注释蛋白质结构域和基序。然后使用TBtools v1.13245绘制Venn图,以显示使用上述三种策略注释的独特和共享蛋白质编码基因。
单倍型组装之间的比较
使用SyRI(Synteny and Rearrangement Identifier)v1.646检测两个单倍型之间的共线性和基因组结构变异(大小≥50 bp),使用默认参数。我们的分析共鉴定出3,011个共线性区域(约350 Mb)、768个易位(约45 Mb)、20个倒位(约2 Mb)、单倍型A中的2,175个重复(约15 Mb)和单倍型B中的1,686个重复(约8 Mb)。大多数重复在染色体4和8上发现,大多数倒位在染色体7上发现(图3b)。SyRI v1.6还用于识别SNP、小插入缺失(小于50 bp的插入和缺失)和串联重复。最终,鉴定出1,264,264个SNP(约1 Mb)、105,563个插入(单倍型B中约2 Mb)、100,073个缺失(单倍型A中约2 Mb)和282个串联重复(约1 Mb)。
数据记录
BGI短读段、PacBio HiFi长读段、Hi-C读段和Iso-Seq数据已存储在NCBI(国家生物信息中心)的Sequence Read Archive数据库中,登录号为SRR25456891-SRR2545689447,48,49,50。最终基因组组装已存储在GenBank数据库中,登录号为GCA_030664735.151和GCA_030664755.152。基因组注释可从Figshare存储库获得53。针对该基因组训练和优化的AUGUSTUS模型及MAKER的配置文件可从Figshare存储库获得54。
技术验证
我们首先计算了映射率作为组装准确性的衡量标准。使用BWA-MEM v0.7.17-r118855和Minimap2 v2.2431(默认参数)分别将短读段和长读段重新映射到组装上。在过滤掉非主要比对后计算映射率。总的来说,99.89%的HiFi读段、97.75%的iso-seq读段和99.81%的短读段被映射(表5)。此外,短读段和长读段数据的读覆盖深度在每个分相染色体上均匀分布,表明我们的单倍型解析组装具有高质量(图S1)。
Summary of mapping rates.
| Data set | Reads mapped | Bases mapped | ≥1× | ≥5× | ≥10× | ≥20× |
|---|---|---|---|---|---|---|
| HiFi reads | 99.89% | 99.88% | 99.99% | 99.79% | 96.46% | 32.19% |
| Iso-Seq reads | 97.75% | 99.13% | 20.77% | 11.37% | 8.59% | 6.34% |
| Short reads | 99.81% | 99.81% | 99.97% | 99.89% | 99.73% | 98.52% |
我们使用BUSCO(Benchmarking Universal Single-Copy Orthologs)v5.3.256基于embryophyta_odb10直系同源数据库评估了基因组组装的完整性。对单倍型A的BUSCO评估鉴定出1,591个完整的BUSCO(包括1,561个单拷贝和30个重复的BUSCO),占单倍型的98.6%,而缺失的BUSCO仅占0.7%(表6)。类似地,对单倍型B的BUSCO评估鉴定出1,588个完整的BUSCO(包括1,560个单拷贝和28个重复的BUSCO),占单倍型的98.4%,而缺失的BUSCO仅占0.9%(表6)。这表明组装相对完整。我们使用Merqury v1.357估计了基因组组装的一致性和完整性。我们的结果显示基因组组装的一致性质量值(QV)为73.38,完整性值为99.19%(表6)。我们还使用KAT(K-mer Analysis Toolkit)v2.4.058通过比较HiFi读段和组装中的k-mers估计基因组组装的质量。结果显示读段和基因组组装之间的一致性很高(图4a),每个单倍型分别代表约一半的杂合峰和几乎所有的纯合峰(图4b,c)。
Evaluation of M. paniculata genome assembly.
| Program | Library | Haplotype A | Haplotype B | Genome |
|---|---|---|---|---|
| BUSCO | Complete BUSCOs (C) | 1,591/98.6% | 1,588/98.4% | 1,591/98.6% |
| Complete and single-copy BUSCOs (S) | 1,561/96.7% | 1,560/96.7% | 9/0.6% | |
| Complete and duplicated BUSCOs (D) | 30/1.9% | 28/1.7% | 1,582/98.0% | |
| Fragmented BUSCOs (F) | 11/0.7% | 12/0.7% | 11/0.7% | |
| Missing BUSCOs (M) | 12/0.7% | 14/0.9% | 12/0.7% | |
| Total BUSCO groups searched | 1,614 | 1,614 | 1,614 | |
| Merqury | Consensus quality value (QV) | — | — | 73.38 |
| Completeness | — | — | 99.19% |
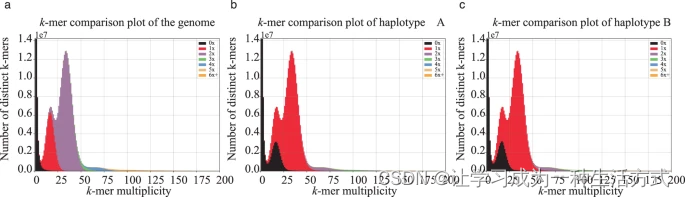
使用KAT(K-mer Analysis Toolkit)绘制的基因组(a)、单倍型A(b)和单倍型B(c)的拷贝数谱图。来自HiFi读段的k-mers显示两个主要的杂合(多重性=18)和纯合(多重性=34)峰,而来自组装的k-mers显示0-6倍的拷贝数。
此外,我们使用BUSCO通过仅保留每个基因的最长蛋白质序列来评估基因组注释的完整性,发现单倍型A的注释为97.6%完整,仅缺失17个(1.1%)基因,单倍型B的注释为97.1%完整,仅缺失19个(1.2%)基因(表7),这表明注释质量很高。
BUSCO evaluation of M. paniculata genome annotation.
| Library | Haplotype A | Haplotype B | Genome |
|---|---|---|---|
| Complete BUSCOs (C) | 1,576/97.6% | 1,567/97.1% | 1,591/98.5% |
| Complete and single-copy BUSCOs (S) | 1,553/96.2% | 1,541/95.5% | 75/4.6% |
| Complete and duplicated BUSCOs (D) | 23/1.4% | 26/1.6% | 1,516/93.9% |
| Fragmented BUSCOs (F) | 21/1.3% | 28/1.7% | 9/0.6% |
| Missing BUSCOs (M) | 17/1.1% | 19/1.2% | 14/0.9% |
| Total BUSCO groups searched | 1,614 | 1,614 | 1,614 |
使用Juicer v1.5.610将Hi-C读段对齐到基因组组装,使用默认参数。使用Juicebox12工具的pre命令(pre -n -q 0或1)将Juicer生成的原始文件转换为hic格式,并使用dump命令(dump observed BP 100000)从hic文件中提取100 kb的接触矩阵。使用Juicebox可视化hic文件。在伪染色体的对角线上观察到强烈的交互信号,对角线外没有明显噪音(图5a),表明该染色体组装的高质量。此外,在排除重复读段后,未在每对同源染色体上观察到异常(图5b),这表明在分相单倍型之间没有切换错误。
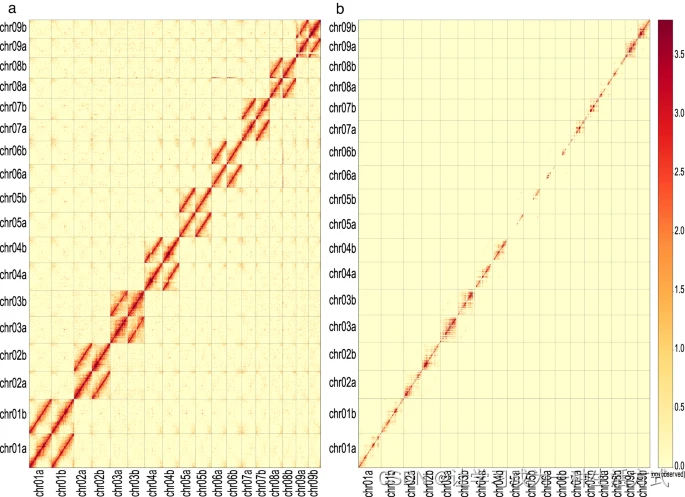
单倍型A和单倍型B的Hi-C交互热图,读段比对质量≥0(包括重复读段)(a) 和比对质量≥1(不包括重复读段)(b)。颜色条表示交互强度,黄色代表低强度,红色代表高强度。
Code availability
All commands and pipelines used were performed according to the manuals or protocols of the tools used in this study. The software and tools used are publicly accessible, with the version and parameters specified in the Methods section. If no detailed parameters were mentioned, default parameters were used. No custom code was used in this study.
这篇关于破布叶(Microcos paniculata)单倍型染色体级别基因组-文献精读22的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!