samtools专题

生信技能57 - Samtools获取指定外显子区域depth和提取BAM文件序列
1. Samtools depth 根据指定bed文件,获取指定区域的覆盖度信息。 # 提取IDT xGen V1 HBA1 exon bedcat xgen-exome-hyb-panel-v1-targets-hg19.bed|grep -w HBA1 > hba.exon.bed# 提取HBA1 外显子的覆盖度# -b: 提取depth的bed文件samtools depth -b

生信软件 | Samtools(SAM文件处理工具)
介绍 SAM(sequence Alignment/mapping) 数据格式是目前高通量测序中存放比对数据的标准格式转换 BAM 与 SAM 格式比对文件排序,建立fastq索引 安装 conda install -y samtools 这里需要安装Conda (这是一款用于安装多数生物信息分析软件的管理软件,重要的是可以解决软件依赖问题) : Conda 安装使用图文详解 使用

生信学习笔记:用conda安装bwa、samtools和tophat2以及问题解决
用conda安装bwa、samtools和tophat2 bwa $ conda install bwa samtools $ conda install samtools tophat2 安装 wget http://ccb.jhu.edu/software/tophat/downloads/tophat-2.1.0.Linux_x86_64.tar.gz 解压 tar -zxvf

sambamba — samtools 的高效平替工具
sambamba — samtools 的高效平替工具 sambamba 是一个 BAM 文件处理工具。 sambamba 它使用了 D 语言的多线程和异步 IO 特性,实现了高效的并行化处理。sambamba 可以在多核 CPU 上同时运行多个任务,利用硬盘和内存的带宽,提高了处理速度。sambamba 还使用了一些优化算法和数据结构,比如快速排序,哈希表,位图等,减少了内存占用和磁

samtools用法详解
文章目录 下载安装测试数据命令详解dictfaidxindexreheaderrmdupcatmergempileup查看参数mpileup生成的结果有参考序列的pileup使用生成一个简单的vcf文件 sort查看用法主要参数释义: splitfastqfastabedcovdepthflagstatidxstatsstatsflagstviewviewbam文件转换为sam文件sam文件

samtools能在windows运行吗?
samtools是ngs分析流程中常用的工具,其主要依赖于zlib,gcc,g++ 和clang,所以直接在windows运用不太可能。那么在类UNIX环境下能不能装好并运行呢。 这里用了Cygwin来进行安装。 Cygwin 安装 Cygwin安装教程【超详细】 samtools samtools 和 bcftools 都是依赖于htslib, 先看下htslib的依赖。 由于Cygwin 安
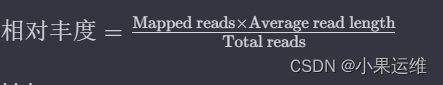
基于BWA,Bowtie2,samtools、checkm等工具计算宏基因组学序列分析中Contigs与Genes在样品中的丰度,多种计算方式和脚本对比
计算contigs和genes相对丰度可以提供有关微生物群落结构和功能的信息。以下是计算这两个指标的意义: 1. Contigs的相对丰度:contigs是利用基因组测序技术获得的碎片序列,通过计算contigs的相对丰度可以了解微生物群落中不同菌种的相对丰度。这可以帮助研究者理解微生物群落的物种组成和群落结构。 2. Genes的相对丰度:基因是生物体内功能的基本单位,通过计算基因的相对丰度

samtools及bam文件的相关知识
文章目录 一些重要的概念首先是关于模板和read的概念然后是线性比对(linear alignment)和嵌合比对(chimeric alignment)的概念Phred scale1-base和0-base文件 bam文件的headerbam文件的主要内容 本文主要记录了阅读http://samtools.github.io/hts-specs/SAMv1.pdf 时学习到的一些内