本文主要是介绍生信分析进阶2 - 利用GC含量的Loess回归矫正reads数量,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
在NGS数据比对后,需要矫正GC偏好引起的reads数量误差可用loess回归算法,使用R语言对封装的loess算法实现。
在NIPT中,GC矫正对检测结果准确性非常重要,具体研究参考以下文章。
Noninvasive Prenatal Diagnosis of Fetal Trisomy 18 and Trisomy 13 by Maternal Plasma DNA Sequencing
链接地址:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3130771/
 窗口划分可参考文章:
窗口划分可参考文章:
生信软件8 - bedtools进行窗口划分、窗口GC含量、窗口测序深度和窗口SNP统计
获取参考基因组大小
以hg19参考基因组为例。
# 安装python库
pip install pyfaidx# 保留chr1-chr22 chrX chrY
faidx reference/hg19.fasta -i chromsizes|grep -E -v '_|chrM' > hg19.genome.size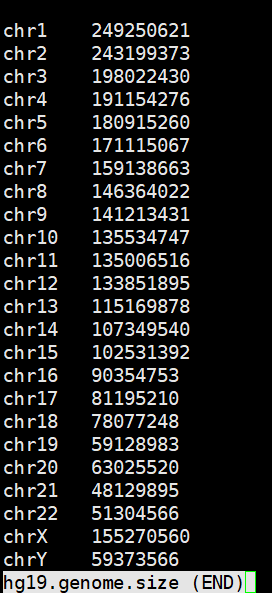
划分基因组窗口
以1000kb划分为例。
bedtools makewindows -g hg19.genome.size -w 1000000 > hg19.1000kb.bed
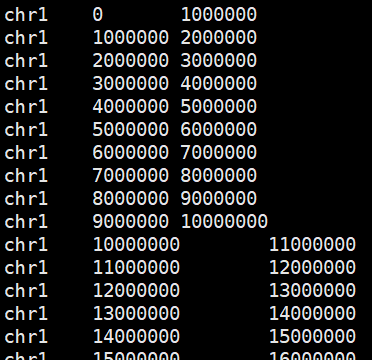
划分窗口
bedtools nuc -fi /reference/hg19.fasta -bed hg19.1000kb.bed|cut -f 1-3,5 > hg19.1000kb.gc.bed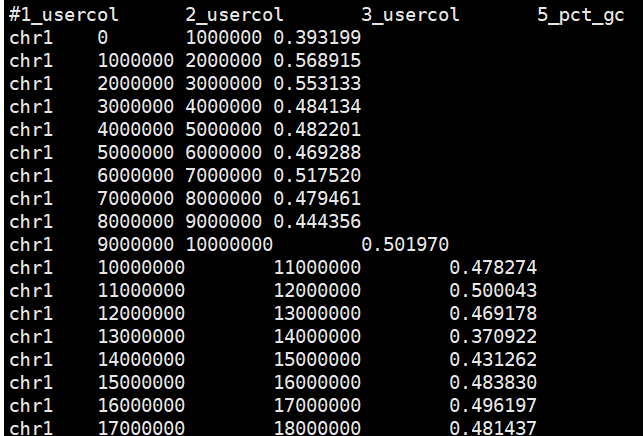
统计窗口reads和GC含量
bedtools coverage -a hg19.1000kb.bed -b sample.sorted.bam > sample.count

整理数据
paste <(grep -v '#' hg19.1000kb.gc.bed) <(cut -f4 sample.count)|sed '1i chr\tstart\tend\tGC\treads' > sample.gc.count
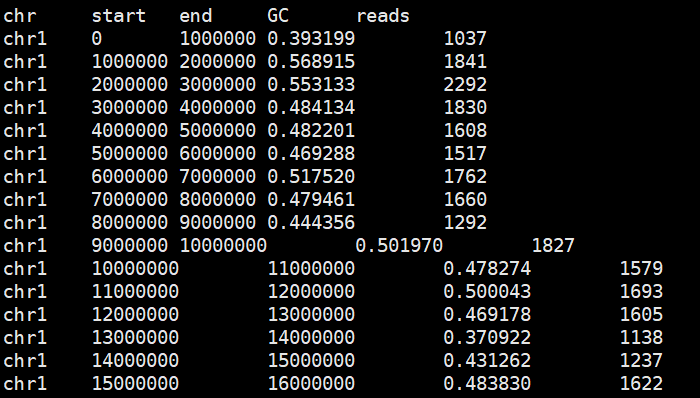
利用GC含量的Loess回归矫正reads数量
R语言实现。
# loess_gc_correct.R
# Useage: Rscript loess_gc_correct.R /path/sample.gc.count /path/sample.gc.corrected.countargs=commandArgs(T)
# 输入文件路径
gc.reads.file <- args[1]
# 输出文件路径
gc.reads.corrected.file <- args[2]# 读取输入文件
raw.data <- read.table(gc.reads.file, sep='\t', head=TRUE)# loess regression 进行GC矫正reads数量
gc.count.loess <- loess(reads~GC,data = raw.data,control = loess.control(surface = "direct"), degree=2) prediction <- predict(gc.count.loess, raw.data$GC)raw.data$corrected_reads <- as.integer(prediction)# 保存
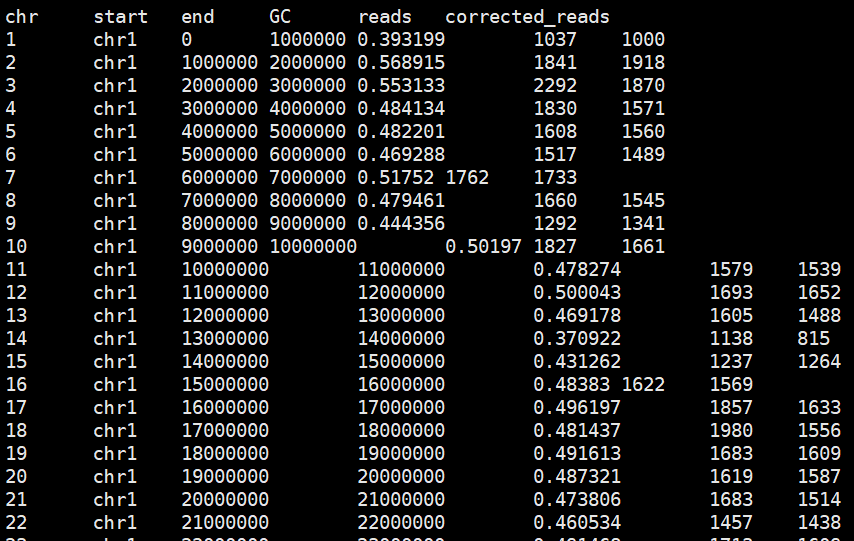
write.table(raw.data, file = gc.reads.corrected.file,sep = '\t', quote = FALSE)矫正后结果
这篇关于生信分析进阶2 - 利用GC含量的Loess回归矫正reads数量的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!






