本文主要是介绍Nature | 通过全球宏基因组学揭示功能性暗物质,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
今天为大家介绍的是来自Nikos C. Kyrpides团队的一篇论文。宏基因组包含了海量多样的蛋白质序列,反映了多种功能和活性。过去,我们通常通过将宏基因组中的序列与参考微生物基因组和那些基因组衍生的蛋白质家族进行比较分析,从而探索这些序列空间。然而,这种方法的局限性在于它只能探索已知的、与参考基因组相关的功能多样性。为了突破这一局限,探索更多未知的功能多样性,作者开发了一种计算方法,可以在不依赖参考基因组的情况下,从宏基因组的序列空间中生成蛋白质家族。

宏基因组的Shotgun测序已经成为研究和分类各种生态环境中微生物的首选方法。随着全基因组测序技术的最新进展以及质量和成本效益的不断提升,大规模测序变得越来越容易、快速。这导致过去几年宏基因组测序数据的数量显著增加,使其成为研究微生物暗物质的不可或缺的资源。为了阐明宏基因组样本的遗传组成,通常有两种主要的方法,每种方法都有明显的优势和劣势。在第一种方法中,测序读数被准确地映射到一个已知的、有注释的参考基因组序列集,以快速了解已知生物、基因和潜在功能的存在。在第二种方法中,可以把测序得到的一连串碱基信息通过de novo(无需参照基因组)组装的方式,拼接成连续的DNA片段,称为contigs或scaffolds,这种组装的过程可以为我们揭示一些以前没有被描述过的微生物及其基因组成的信息。然而,这两种方法在基因功能注释方面都存在同样的限制,即依赖于通过同源搜索来预测蛋白质数据库中的功能。因此,映射不到参考蛋白质家族的基因通常会被忽略,并从后续的比较分析中丢弃。为了消除对参考数据集的依赖,以估计未探索的功能多样性的广度(作者称其为功能性暗物质),需要进行全面的宏基因组比较。但这项任务需要大量的计算资源,达到这种可扩展性仍然具有技术挑战。尽管最近有一些优秀的努力来解决这个问题,但宏基因组尚未全面调查以揭示功能性暗物质。在这里,作者提出了一种可扩展的计算方法,用于识别和描述在宏基因组中发现的功能性暗物质。作者首先在IMG/M的26,931个宏基因组数据集中识别了新的蛋白质空间,去除了与IMG数据库中的超过100,000个参考基因组或Pfam匹配的所有基因。然后将剩余的序列聚类成蛋白质家族,并探索了它们的分类学和生态分布,以及在可能的情况下,预测它们的三级(三维(3D))结构。
新的蛋白质序列空间
首先,作者从IMG/M平台上所有公开的参考基因组、宏基因组和宏转录组中收集了所有长度超过35个氨基酸残基的蛋白质序列。总共从89,412个细菌基因组、9,202个病毒基因组、3,073个古菌基因组和804个真核生物基因组中提取了所有的蛋白质序列,得到了最终的数据集,共计94,672,003个序列。研究包括的参考基因组仅包括分离基因组,不包括宏基因组和单独扩增的基因组。同样,对于未分类的宏基因组,作者从至少500 bp的scaffolds中提取了所有预测的蛋白质序列,这些序列的长度至少为35个氨基酸,共计26,931个数据集(20,759个宏基因组和6,172个宏转录组),作者将其称为环境数据集(ED)。这样得到了一个非冗余的数据集,共计8,364,611,943个预测的蛋白质或蛋白质片段。为了识别这个数据集中的功能性暗物质成分,作者首先舍弃了任何与Pfam数据库中的序列或参考基因组集中的序列匹配的蛋白质序列。最后,代表未探索的宏基因组蛋白质空间的非冗余目录包括1,171,974,849个蛋白质序列(占总数的14%)。
新蛋白质家族
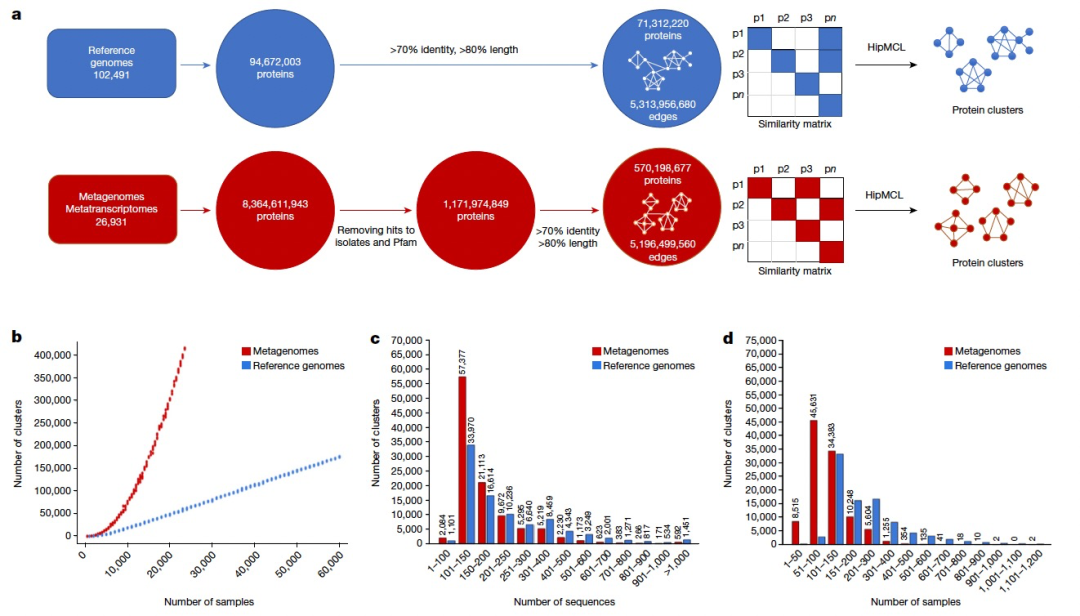
接下来,作者对11亿ED蛋白质进行了聚类。为了进行比较,作者对来自参考基因组的9400万蛋白质也采用了相同的方法。首先为这两个基因目录(即来自参考基因组的蛋白质和来自ED的蛋白质)分别构建了一个所有对所有的相似性矩阵,通过计算所有显著的序列相似性对。然后,分析这两个图以识别基于序列相似性的蛋白质簇。为此,作者使用了HipMCL,这是原始MCL算法的大规模并行实现。从数据检索到簇生成的整个过程如图1a所示。
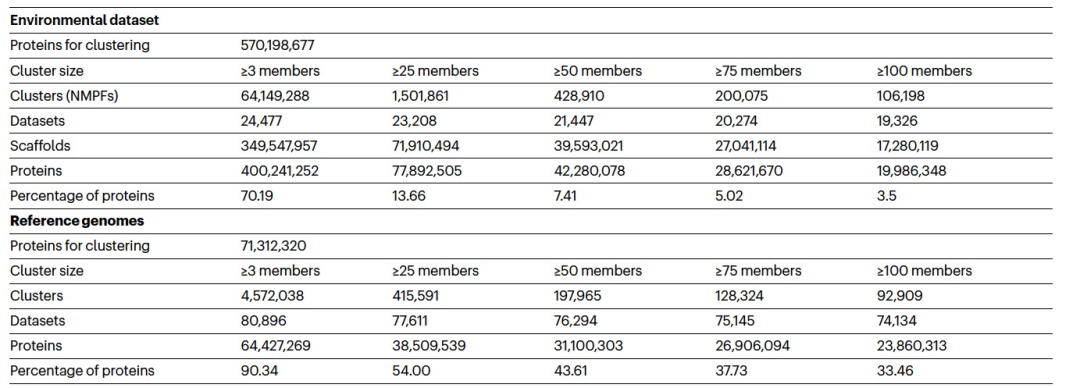
虽然至少有50个成员的簇(甚至可能是至少有25个成员的簇)可能代表潜在的功能重要簇,但作者将后续分析限制在至少有100个成员的较大家族上,以便专注于更高质量的数据,以及更好的预测结构的候选对象(表1)。总共,作者识别了至少有100个成员的106,198个家族,它们将被称为新的宏基因组蛋白质家族(NMPFs)(表1(右列))。作为比较,作者在相应的参考基因组蛋白质簇中识别了92,909个蛋白质簇。通过直接比较这两组簇(参考簇与ED蛋白质簇),可以观察到,对于至少有3个成员的簇,ED蛋白质簇增加了14倍以上;对于至少有25个成员的簇,增加了3倍以上;对于至少有50和75个成员的簇,增加了约2倍;对于至少有100个成员的簇,也有所增加(表1)。虽然与参考基因组相比,宏基因组序列空间本质上更碎片化,并且错误或不完整的基因比例更高,但这些结果也表明,仍然有许多蛋白质序列空间有待探索。这也得到了由至少100个成员的簇生成的稀疏曲线(图1b)的支持。这些曲线显示,随着样本数量的增加,参考基因组的簇数量线性增加,但宏基因组的簇数量指数增加,且没有达到平稳状态。
生态系统分布情况
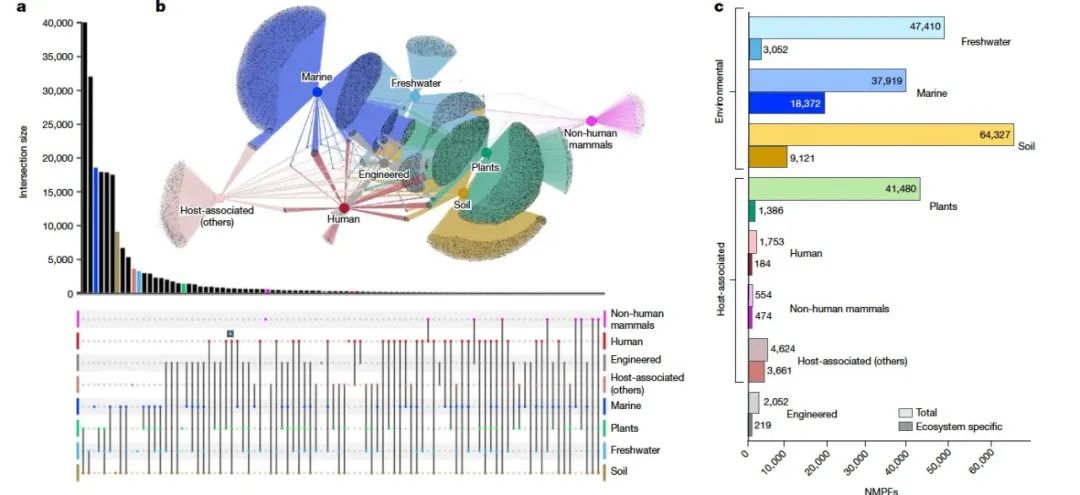
为了确定NMPFs的生态系统分布情况,作者通过IMG/M平台使用GOLD数据库的生态系统分类方案收集了每个样本的相应元数据。NMPFs的生态系统分布如图2a,b。这里的三个主要GOLD生态系统(环境、宿主相关和工程)被进一步划分为八种更具体的生态系统类型:淡水、海洋、土壤、植物、人类、非人类哺乳动物、其他宿主相关和工程。通过观察网络拓扑,可以发现在三个宽泛的生态系统中,每个NMPF的基因共享最少,这与最近从13,174个宏基因组中对蛋白质家族的观察结果一致,土壤/植物关联除外。最多的NMPFs在土壤和植物环境之间共享(土壤的62%和植物相关家族的96%),这是符合预期的,因为这些生态系统的采样重叠较大(图2a)。其次是土壤和淡水之间共享的NMPFs,这主要是由于湿地和沉积物样本被归类为淡水生态系统。同样的原因,可以观察到植物和淡水NMPFs之间以及土壤、淡水和植物NMPFs之间也存在明显的重叠。相反,淡水和海洋NMPFs之间只有37%的共享,人类、非人类哺乳动物和宿主相关生态系统类型之间的蛋白质家族共享更少。另一方面,观察到人类和工程环境之间存在相当大的NMPFs重叠(图2)。这并不令人惊讶,考虑到工程环境主要包含与人类废物相关的生态系统样本(如固体废物和废水)。类似地,淡水和工程环境之间以及淡水和宿主相关类型(人类、非人类哺乳动物和其他宿主相关)之间也存在重叠。这些重叠可能表明现象,例如淡水环境的粪便污染,导致相同的NMPFs和因此相同的微生物群落在不同的生态系统类型中共存。
分类学分布
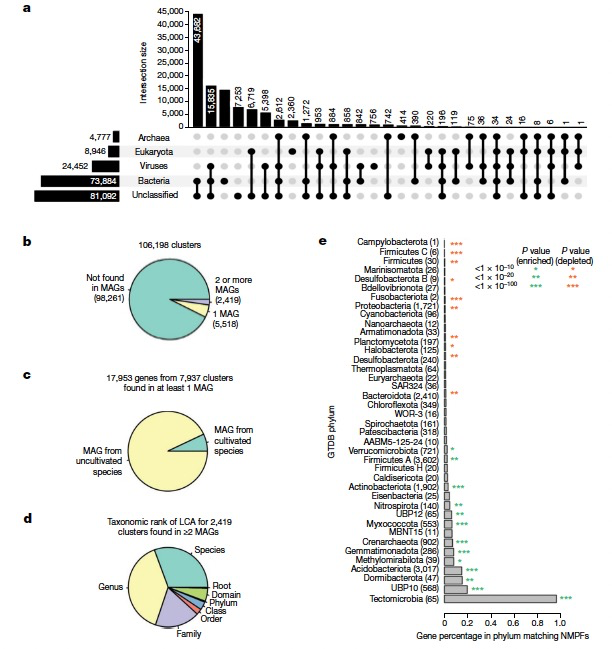
作者根据IMG数据库中相应支架的可用分类学信息,为每个簇中的NMPF成员进行了分类学分配。如果没有这样的注释,作者就用其他的方法组合起来推断支架的分类学。在总共17,280,119个包含NMPF成员的IMG/M支架中,8,049,154个被分类为细菌,382,761个为古菌,1,184,393个为真核生物,1,406,588个为病毒,剩下的6,257,223个为未分类。基于它们相应的支架分类学分配,NMPFs的分类学分布如图3a所示。大多数蛋白质家族包括具有多个分类学分配的序列(例如细菌和未分类,或细菌和病毒)。最大的类别包括具有细菌/未分类序列的家族,其次是病毒/未分类和细菌/病毒。一小群家族被分配给真核生物,更少的家族被分配到古菌。最后,有7,253个簇完全没有分类学信息。接下来,作者评估了在最近识别的地球微生物组(Genomes from Earth's Microbiomes,GEM)目录中,是否有任何NMPF蛋白质(及其相应的家族)被发现。具体来说,作者检查了包含NMPF基因的支架是否在GEM目录的52,515个MAGs中被分档。结果显示,在GEM目录中发现了17,953个基因,来自7,937个NMPF(占总数的7.4%)(图3b,c),其中绝大多数(93%)来自未培养的物种。对于那些在两个或多个MAGs中出现的家族,可以注意到它们在分类学上有一个强烈的狭窄分布,超过三分之二的家族被限制在单一的物种或属,只有极少数家族分布在多个科、目或门中(图3d)。发现NMPF在土壤环境中常见的几个门中富集(例如,Gemmatimonadota、Acidobacteriota、Crenarchaeota和Myxococcota),而在人类和其他宿主相关的环境中所发现的几个门中则较少(Firmicutes、Proteobacteria和Bacteroidota;图3e)。总之,这些结果揭示了尽管在整个过程中和大规模MAG重建中取得了改进,但仍有相当一部分功能多样性在分类学上保持孤立。
元数据分布
接下来,作者检查了NMPFs的地理分布。极少数的家族(1,372个,占1.3%)被发现具有有限的地理分布(在1公里内),当允许的最大距离为1,000公里时,这个数字只是适度增加(4,330个,占4%)。这些家族中的大多数被发现在植物、土壤和淡水生态系统中。其中极少数的家族包括在海洋生态系统或人类样本中发现的成员,这与这些生态系统中较高的微生物扩散是一致的。大多数NMPFs(64,186个或60.44%)由来自宏基因组和宏转录组的蛋白质混合而成,进一步验证了它们的存在,而38,292个(36.06%)的NMPFs包含了仅在宏基因组中发现的蛋白质,3,720个(3.50%)的NMPFs包含了仅在宏转录组中发现的蛋白质。随着家族成员数量的减少,包含来自宏基因组和宏转录组成员的家族百分比逐渐减少。同时,这些在宏基因组和宏转录组中都发现的NMPFs也有最广泛的样本分布,即在最多的样本中发现了这些家族。这些家族的大多数被分类为环境生态系统(主要是土壤,较少的是海洋和淡水样本),并主要包含细菌和未分类的序列。为了估算新蛋白质家族在环境测序数据中的分布,作者将从每个支架提取出来并用于本研究的新蛋白质的数量与相应支架中的总基因/蛋白质数量进行了比较。作则会分析的大多数支架(13,407,728个,占77.59%)都包含了新的和已知的基因。新的与这些支架中的总基因数量的比较显示,支架的大小或每个支架的总基因数量与新基因的数量没有相关性。作者研究中的最大支架(4,302个基因)只包含了一个新序列。通常情况下,研究中最大的支架只包含了有限数量的新序列,并且源自细菌或未分类的宏基因组样本。相反,包含最多新序列的支架长度(和基因数量)变化较大,且大多数源自病毒。
结构分布
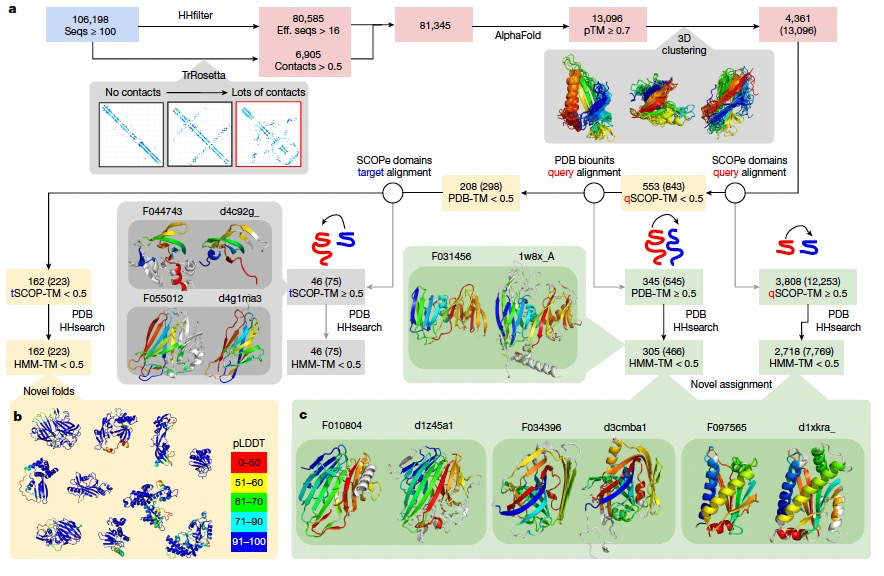
近期在蛋白质结构预测领域的突破使得对蛋白质序列进行快速且准确的结构表征成为可能。已有研究表明,宏基因组序列是发现新结构的一个特别丰富的源头。在这里,作者运用AlphaFold2对至少包含16个多样化序列的NMPFs进行分析,或者对TrRosetta预测出的结构良好的蛋白质进行分析。结果总结如图4a所示。在符合上述条件的81,345个NMPFs中,预测出了80,585个3D模型,其中13,096个NMPFs的预测置信度较高(预测的TM(pTM)分数 > 0.700)。基于结构聚类,这些高置信度的预测代表了4,361个独特的结构。为了检查这些结构的新颖性或功能,作者将它们与SCOP-Extended(SCOPe)中的实验确定结构和PDB中的复合物进行比较。总共有3,808个结构(包含12,253个NMPFs)与至少1个SCOPe域有显著的结构重叠(TM分数 > 0.5)。其中,2,718个(包含7,769个NMPFs)有高的命中,表示62.3%的高质量预测至少与一个SCOPe域或PDB中的复合物有一定的相似性。现在,这些基于结构相似性的新分配可以用于预测相应序列的功能。图4c展示了一些示例。例如,家族F034396在使用HHsearch与PDB进行比对时没有命中,但在使用结构搜索与SCOPe域进行比对时,它有一个强的命中。作者强调,这些情况应被视为有根据的预测,需要通过实验进行验证,因为相同的折叠不总是对应相同的功能。然而,可以通过将这些新分配与其他NMPF元数据(如基因共生现象)结合起来,来进行一些验证和额外的功能注释。为了确认剩余的553个没有SCOPe命中的蛋白质是新的折叠,作者对所有PDB生物中的复合物进行了更为详尽的搜索,包括所有可能的链排列。总共有345个模型命中了至少一个PDB条目,其中305个代表了额外的新分配。剩余的208个被处理以进行进一步的过滤,去除了50%的结构与一个SCOPe域匹配的预测。最终,从223个NMPFs中识别出了162个折叠和/或域-域定向被认为是新颖的(图4b)。
参考资料
Pavlopoulos, G.A., Baltoumas, F.A., Liu, S. et al. Unraveling the functional dark matter through global metagenomics. Nature 622, 594–602 (2023).
https://doi.org/10.1038/s41586-023-06583-7
这篇关于Nature | 通过全球宏基因组学揭示功能性暗物质的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!

![[机缘参悟-222] - 系统的重构源于被动的痛苦、源于主动的精进、源于进化与演进(软件系统、思维方式、亲密关系、企业系统、商业价值链、中国社会、全球)](https://i-blog.csdnimg.cn/direct/fd1df13932fb4df09c34297f62f78bf0.png)






