本文主要是介绍scATAC-seq(Signic)官方分析流程,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
Analyzing PBMC scATAC-seq(官方教程 )
分析由10x Genomics提供的人类外周血单核细胞(PBMCs)的单细胞ATAC-seq数据集
一、Signac的安装 & 加载流程中所有的安装包
install.packages("Signac")
install.packages("BiocManager")
install.packages("EnsDb.Hsapiens.v75")
install.packages("Seurat")
install.packages("spatstat.utils")
install.packages("spatstat.geom", dependencies = TRUE)
install.packages("hdf5r")
BiocManager::install("biovizBase")library(EnsDb.Hsapiens.v75) ##这个包用于人类的基因组注释文件
library(patchwork)
library(Signac)
library(Seurat)
library(ggplot2)
library(hdf5r)
library(spatstat.geom)
library(biovizBase)
二、导入数据 创建Signic对象
输入文件介绍
Signac需要两个输入文件,这两个输入文件都可以由CellRanger获得
(1)Peak/Cell matrix:
横坐标为每个peak区域,纵坐标为每个细胞。类似于单细胞的gene expression count matrix
每个peak区域代表预测的每一个染色质开放区域
矩阵内的数值代表该细胞在这个个peak位置中Tn5酶结合的个数
(2)Fragment file:
该文件为一个列表,其中包含了所有单细胞中的所有的unique—fragement
这个文件很大,通常储存在磁盘(disk)上,而不需要导入内存(memory)中
这个文件的作用就是展示一个细胞中所有的fragment片段,而不是仅包含peak里面的片段
setwd("D:/R/Signac.pipeline/data") ##设置工作路径# 读入CSV描述数据属性文件到metadata
metadata <- read.csv(file = "D:/R/Signac.pipeline/data/atac_v1_pbmc_10k_singlecell.csv",header = TRUE,row.names = 1
) # 读入Peak/Cell matrix
counts <- Read10X_h5(filename = "D:/R/Signac.pipeline/data/atac_v1_pbmc_10k_filtered_peak_bc_matrix.h5")# ATAC-seq的数据(peak和fragment)被整合并储存在一种特殊的assays(ChromatinAssay)
chrom_assay <- CreateChromatinAssay(counts = counts,sep = c(":", "-"),genome = 'hg19',fragments = 'D:/R/Signac.pipeline/data/atac_v1_pbmc_10k_fragments.tsv.gz',min.cells = 10,min.features = 200
)# 将(ChromatinAssay)创建为seurat对象
pbmc <- CreateSeuratObject(counts = chrom_assay,assay = "peaks",meta.data = metadata
)#通过打印这个peaks的assays,可看其中的附加信息motif information、 gene annotations、genome information
pbmc[['peaks']]#可以使用grangs函数应用于Seurat对象 或者 “pbmc@assays$peaks@ranges”直接获取每个peaks在基因组上的ranges(范围)
granges(pbmc)pbmc@assays$peaks@ranges
GRanges object with 87561 ranges and 0 metadata columns:
seqnames ranges strand
[1] chr1 565107-565550 *
[2] chr1 569174-569639 *
[3] chr1 713460-714823 *
[4] chr1 752422-753038 *
[5] chr1 762106-763359 *
… … … … [87557] chrY 58993392-58993760 * [87558] chrY 58994571-58994823 *
[87559] chrY 58996352-58997331 * [87560] chrY
59001782-59002175 * [87561] chrY 59017143-59017246 *
------- seqinfo: 24 sequences from an unspecified genome; no seqlengths
三、pbmc对象中添加人类基因组的gene的注释
# 我们可以向pbmc对象中添加人类基因组的gene的注释,下游函数(FUNCTION)就可以直接从seurat对象中提取gene的注释用于分析(如,画track)
BiocManager::install("AnnotationHub")
library(AnnotationHub)hub <- AnnotationHub()
query(hub, c("Homo sapiens","ensdb"))##检索ensdb数据中人类基因组
homo_ensdb <- hub[["AH53211"]]##提取某个注释文件#library(EnsDb.Hsapiens.v75)
#从Ensb包中获取hg19版本人基因组注释
annotations <- GetGRangesFromEnsDb(ensdb = EnsDb.Hsapiens.v75)
#将注释文件由EnsDb格式转换为GRanges格式
seqlevelsStyle(annotations) <- "UCSC"
#给pbmc对象设置注释文件
Annotation(pbmc) <- annotations
四、Computing QC Metrics
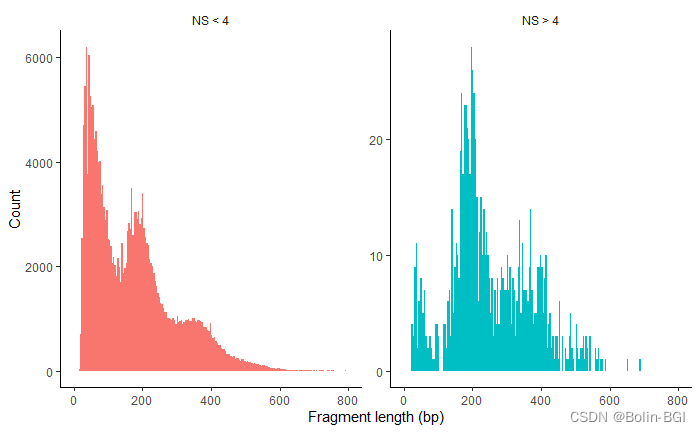
核小体条带分布模式(Nucleosome banding pattern)————Signic独有
# compute nucleosome signal score per cell
pbmc <- NucleosomeSignal(object = pbmc)
pbmc$nucleosome_group <- ifelse(pbmc$nucleosome_signal > 4, 'NS > 4', 'NS < 4')
FragmentHistogram(object = pbmc, group.by = 'nucleosome_group')
#可见,单核小体/无核小体比率(the mononucleosomal / nucleosome-free ratio)的异常值细胞具有不同的核小体条带模式
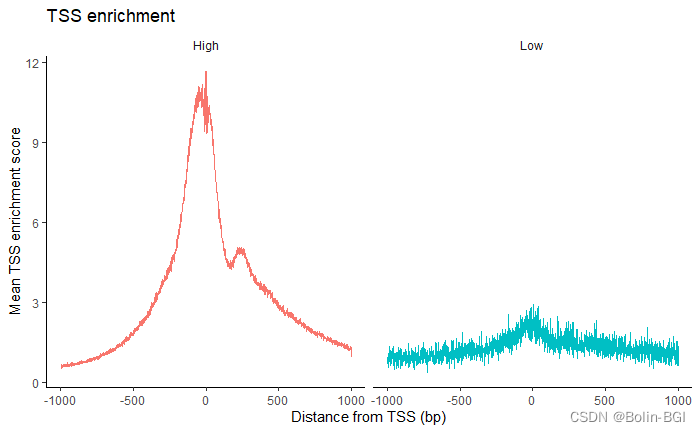
转录起始位点富集分数(Transcriptional start site (TSS) enrichment score)
# compute Transcriptional start site (TSS) enrichment score
pbmc <- TSSEnrichment(object = pbmc, fast = FALSE)
# 当fast=T时,仅计算TSS分数,而不存储每个细胞Tn5插入频率的位置矩阵,这样后续将不能使用TSSPlot画图。
# 基于TSS分数将不同的细胞进行分组以及画所有TSS位点的可及性信号图来检查TSS富集分数
pbmc$high.tss <- ifelse(pbmc$TSS.enrichment > 2, 'High', 'Low')
TSSPlot(pbmc, group.by = 'high.tss') + NoLegend()
pbmc$pct_reads_in_peaks <- pbmc$peak_region_fragments / pbmc$passed_filters * 100 #将peaks中reads的比例加进来
pbmc$blacklist_ratio <- pbmc$blacklist_region_fragments / pbmc$peak_region_fragments #将blacklist区域中reads比例加进来##画上述5个指标的小提琴图
VlnPlot(object = pbmc,features = c('pct_reads_in_peaks', 'peak_region_fragments','TSS.enrichment', 'blacklist_ratio', 'nucleosome_signal'),pt.size = 0.1,ncol = 5
)
# pct_reads_in_peaks: (reads富集到peaks区域的百分比)是指所有测序reads中被分配到基因组区域中ATAC-seq峰的百分比。pct_reads_in_peaks的计算可以帮助评估ATAC-seq测序数据的质量和信噪比,因为在良好的实验和数据处理条件下,ATAC-seq数据中大部分的reads应该能够映射到ATAC-seq峰的区域中。较高的pct_reads_in_peaks通常表示ATAC-seq测序数据较为可靠,但也可能与样本中存在更多开放的区域相关。# peak_region_fragments:每个ATAC-seq峰区域内包含的ATAC-seq测序片段数。每个峰表示基因组上的一段区域,其相对于周围区域具有更高的ATAC-seq信号。较高的peak_region_fragments通常表示较高的ATAC-seq数据信噪比和较高的峰质量。在一些情况下,peak_region_fragments也可以用于比较不同样本或不同实验之间的差异性和一致性。# TSS.enrichment:(转录起始位点富集度)是指在基因组上具有转录起始位点(TSS)的区域在scATAC-seq数据中出现的比例,与在基因组上随机区域的出现比例相比较得出。转录起始位点(TSS)是指基因的起始位置,是转录调控和基因表达的重要区域。在scATAC-seq中,对每个单细胞的ATAC-seq信号进行peak calling后,可以使用一系列方法来评估每个细胞的TSS富集度,从而鉴定细胞中的基因表达和调控元件。TSS.enrichment是衡量一个细胞是否富集TSS区域的一个指标,通常情况下,高TSS.enrichment值的细胞往往与较高的基因表达水平相关联,因此可以用来鉴定不同细胞类型和状态的基因表达和调控元件。# blacklist_ratio:(黑名单比率)是指在ATAC-seq数据分析中,被标记为"黑名单"的染色质区域所占的比例。黑名单是指染色质上的一些区域,这些区域往往是高度复杂的重复序列,或者是具有高度变异性的区域,如单倍型差异区域和转座子元件等。这些区域会在ATAC-seq数据的分析过程中被过滤掉,以减少假阳性信号的出现。因此,在scATAC-seq数据分析中,blacklist_ratio可以用来评估每个单细胞ATAC-seq数据的质量,如果一个细胞的blacklist_ratio很高,说明这个细胞的ATAC-seq信号被大量地过滤掉,可能需要进一步的筛选或者数据处理来获得更好的结果。# nucleosome_signal:(核小体信号)是指某个基因组区域在ATAC-seq实验中被核小体所保护的程度。该指标通常是通过计算ATAC-seq测序数据中核小体上下游区域ATAC-seq峰的差异性来评估。ATAC-seq是通过使用转座酶打开染色质,使ATAC-seq测序数据中更容易获得开放染色质区域。但是,由于核小体的存在,染色质某些区域会被核小体所保护,从而阻碍转座酶的结合和DNA的开放。因此,ATAC-seq测序数据中,具有核小体保护的区域通常表现出较低的ATAC-seq信号。ATAC-seq是通过使用转座酶打开染色质,使ATAC-seq测序数据中更容易获得开放染色质区域。但是,由于核小体的存在,染色质某些区域会被核小体所保护,从而阻碍转座酶的结合和DNA的开放。因此,ATAC-seq测序数据中,具有核小体保护的区域通常表现出较低的ATAC-seq信号。
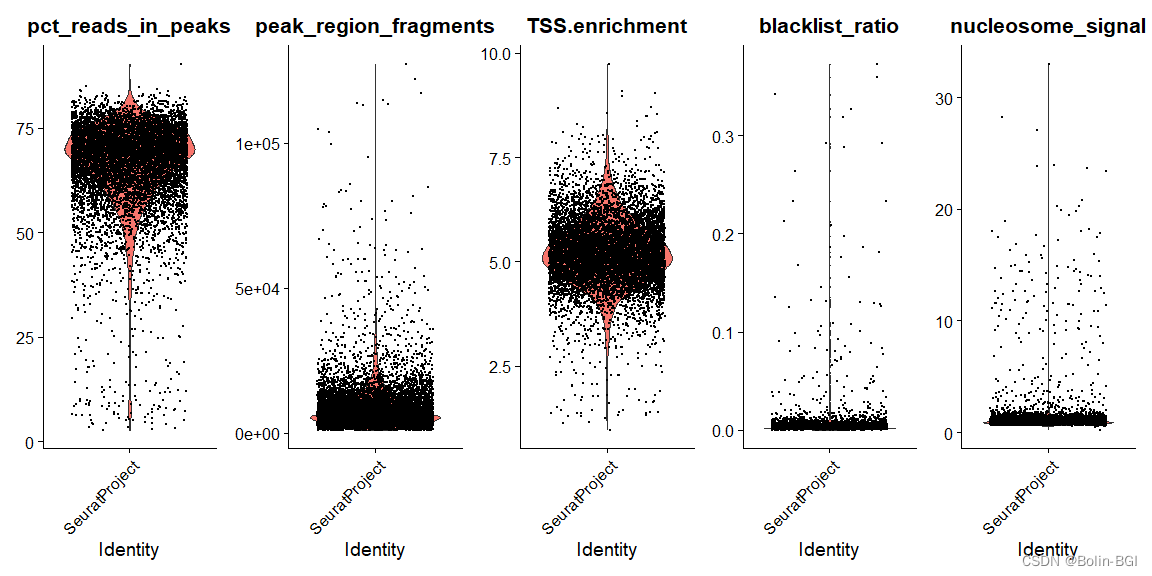
##去除不符合上述5个指标的细胞
pbmc <- subset(x = pbmc,subset = peak_region_fragments > 3000 &peak_region_fragments < 20000 &pct_reads_in_peaks > 15 &blacklist_ratio < 0.05 &nucleosome_signal < 4 &TSS.enrichment > 2
)
pbmc五、标准化及线性降维
标准化/归一化(Normalization)
Signac是基于词频-逆文档频次 (term frequency-inverse document frequency,TF-IDF) 方法进行的归一化。它进行两步(两个维度:横向&纵向)归一化操作,即跨细胞归一化以校正细胞测序深度的差异(可以理解为每个peak在不用cell分布异常的情况),纵向即跨峰(peaks)归一化为更罕见的峰(rare peak)提供更高的值
特征选择 (Feature selection)
由于scATAC-seq的低变化性(因为测的是DNA,所有每个细胞在每个peak能测到的数目要么是1要么是0,最多可能测的在复制的细胞为4),使我们很难像scRNA-seq数据那样筛选高可变特征。作为替代方案,我们仅选择前n%的peaks以进行降维。或者去除仅在少于n个细胞中检测到的peaks(使用FindTopFeatures()函数)在这里我们一般选择降维的方法来加快运行速度(将min.cutoff 设置为‘q75’以使用前25%的peaks )(由于发现使用降维后的子集与全部的peaks得到的结果是相似的)
降维 (Dimension reduction)
接下来对TD-IDF矩阵采用singular value decomposition(SVD)方法进行降维,并使用上一步筛选出的peaks(features),这将返回这个 对象(object)的低维LSI代表(类似于scRNA-seq里面的PCA)这种在TF-IDF后继续进行SVD的方法就是著名的latent semantic indexing (LSI)方法
pbmc <- RunTFIDF(pbmc) # (Normalization)
pbmc <- FindTopFeatures(pbmc, min.cutoff = 'q0 ') # (Feature selection)
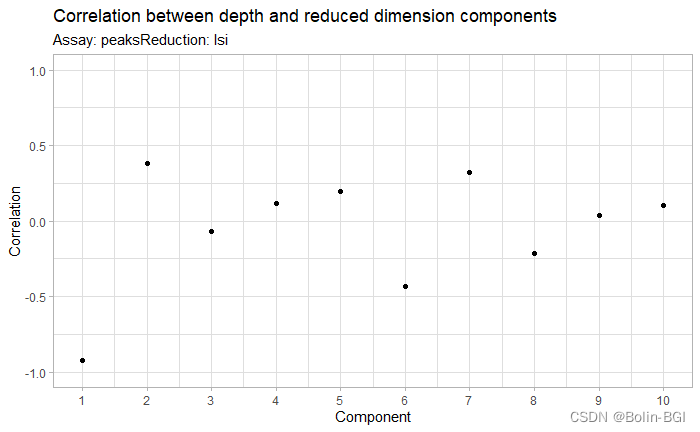
pbmc <- RunSVD(pbmc) # (Dimension reduction)#通常在进行完线性降维后,得到的第一个LSI所代表的通常为技术差异,而非生物学差异,我们一般将第一个LSI去除。
#可以使用DepthCor函数评估每个LSI成分与测序深度之间的相关性DepthCor(pbmc)
# 测序深度和每个降维分量LSI之间的相关性:由图可知,LSI_1与depth之间存在非常强的负相关性,说明LSI_1这个维度反映的是不同细胞之间测序深度之间的差异而不是生物学本身的差异,因此我们去除这个第一个LSI后再执行下游步骤。
六、非线性降维&聚类( Non-linear dimension reduction and clustering)
# 使用 RunUMAP() 函数对 pbmc 对象进行降维操作,其中 reduction 参数指定使用的降维方法为 LSI(Latent Semantic Indexing,隐含语义索引),dims 参数指定保留的维度范围为 2 到 30。
pbmc <- RunUMAP(object = pbmc, reduction = 'lsi', dims = 2:30)
# 使用 FindNeighbors() 函数基于降维后的数据计算样本之间的距离(通常使用欧氏距离或 Pearson 相关系数),并构建一个邻接图,其中 reduction 和 dims 参数与第一行代码相同。
pbmc <- FindNeighbors(object = pbmc, reduction = 'lsi', dims = 2:30)
# 使用 FindClusters() 函数对邻接图进行聚类,其中verbose参数表示是否输出聚类过程的详细信息,algorithm参数表示使用的聚类算法,这里指定为第三种算法(Shared Nearest Neighbor,共享最近邻)。聚类结果将被存储在 pbmc@ident 中。
pbmc <- FindClusters(object = pbmc, verbose = FALSE, algorithm = 3)
# 使用 DimPlot() 函数对聚类结果进行可视化,其中 object 参数指定数据对象为pbmc,label参数表示是否在图上标注聚类结果,NoLegend() 函数表示不显示图例。
DimPlot(object = pbmc, label = TRUE) + NoLegend()
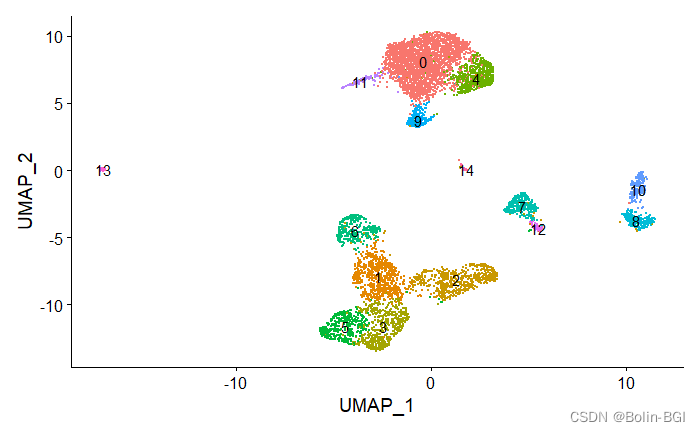
创建基因活性表达矩阵(gene activity matrix)
我们可以通过评估与基因相关的染色质区域的可及性来对基因组中的基因活性进行定量,并根据scATAC-seq数据创建一个新的gene活性表格,我们在这里仅采用一种简单的方法:sum统计与基因体和启动子区域重叠的fragement的个数
为了创建gene活性表达矩阵,我们提取每个gene的坐标并延长至其上游的2kb区域,然后我们统计每个细胞中映射到上述区域的fragement数目,这一步可以使用FeatureMatrix()函数来进行,但是这步已经在 GeneActivity()函数中自动运行
#GeneActivity() 函数自动执行创建基因活性表达矩阵这一步骤
gene.activities <- GeneActivity(pbmc) #将活性矩阵添加到pbmc对象中,作为一个新的名为"RNA"的assays,并进行标准化。
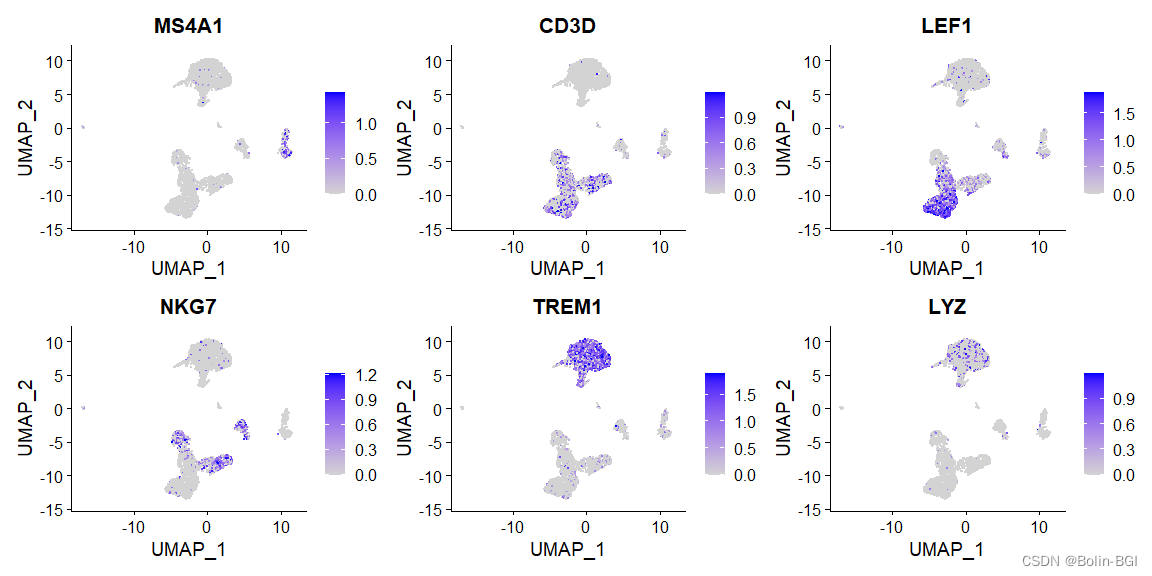
pbmc[['RNA']] <- CreateAssayObject(counts = gene.activities)pbmc <- NormalizeData(object = pbmc,assay = 'RNA',normalization.method = 'LogNormalize',scale.factor = median(pbmc$nCount_RNA))DefaultAssay(pbmc) <- 'RNA'
#使用经典的标记基因(marker genes)的活性值可视化来帮助我们解释ATAC-seq簇。但是这个估计比scRNA-seq数据的估计的噪音值更大,这是因为ATAC-seq所代表的基因活性值与基因的表达不总是相对应的。FeaturePlot(object = pbmc,features = c('MS4A1', 'CD3D', 'LEF1', 'NKG7', 'TREM1', 'LYZ'),pt.size = 0.1,max.cutoff = 'q95',ncol = 3
)
七、与scRNA-seq数据进行整合(Integrating with scRNA-seq data)
为了帮助解释ATAC-seq数据,我们可以基于来自于同一生物学系统的scRNA-seq的数据来对细胞进行分类
我们使用一种标签转移(label transfer)和多组学的数据整合(cross-modality integration)的方法。目的是在基于基因活性矩阵的scATAC-seq数据和基于基因表达的scRNA-seq数据中鉴定到共同的相关模式,匹配两种模态的生物学数据
#加载下载后的PMBC的scRNA-seq的Seurat对象
pbmc_rna <- readRDS("D:/R/Signac.pipeline/data/pbmc_10k_v3.rds")transfer.anchors <- FindTransferAnchors( reference = pbmc_rna,query = pbmc,reduction = 'cca')# 经典关联算法(Classical association algorithm,CCA) 本用于解决批次效应,此处用于整合RNA(reference)和ATAC(query)两个模态的数据,即找到RNA和ATAC之间的Anchors )predicted.labels <- TransferData( #预测的细胞标签anchorset = transfer.anchors,refdata = pbmc_rna$celltype,weight.reduction = pbmc[['lsi']],dims = 2:30
)pbmc <- AddMetaData(object = pbmc, metadata = predicted.labels) #将预测的细胞标签加入pmbc的meta.data中##绘图对两种数据进行可视化
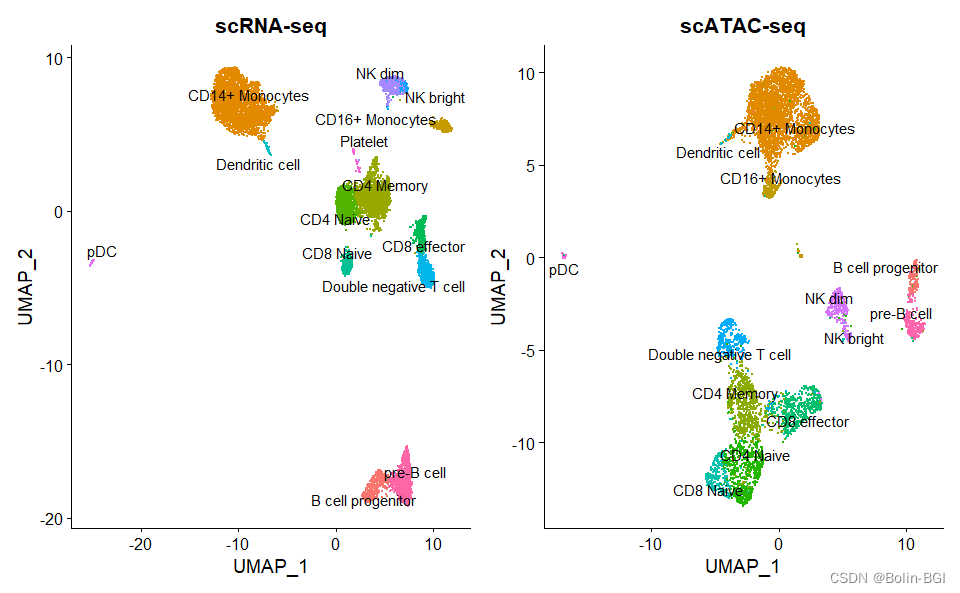
plot1 <- DimPlot(object = pbmc_rna,group.by = 'celltype',label = TRUE,repel = TRUE) + NoLegend() +ggtitle('scRNA-seq')
plot2 <- DimPlot(object = pbmc,group.by = 'predicted.id',label = TRUE,repel = TRUE) + NoLegend() + ggtitle('scATAC-seq')
plot1 + plot2
# 根据图可以看到scRNA-seq的分类与我们仅基于scATAC-seq的数据几乎一致,我们可以很方便的注释scATAC-seq的细胞簇
pbmc <- subset(pbmc, idents = 14, invert = TRUE)
pbmc <- RenameIdents(object = pbmc,'0' = 'CD14 Mono','1' = 'CD4 Memory','2' = 'CD8 Effector','3' = 'CD4 Naive','4' = 'CD14 Mono','5' = 'DN T','6' = 'CD8 Naive','7' = 'NK CD56Dim','8' = 'pre-B','9' = 'CD16 Mono','10' = 'pro-B','11' = 'DC','12' = 'NK CD56bright','13' = 'pDC'
)
八、鉴定不同细胞簇间差异可及性的peaks(Find differentially accessible peaks between clusters)
# 这一步耗时长,我们仅对 Naive CD4 cells 和 CD14 monocytes进行比较查看差异peaks
DefaultAssay(pbmc) <- 'peaks' #将默认的assays转换回peaksda_peaks <- FindMarkers(object = pbmc,ident.1 = "CD4 Naive",ident.2 = "CD14 Mono",test.use = 'LR',latent.vars = 'peak_region_fragments'
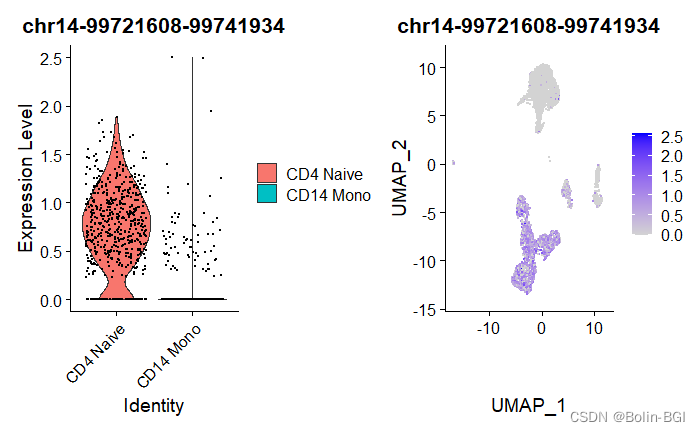
)head(da_peaks)plot1 <- VlnPlot(object = pbmc,features = rownames(da_peaks)[1],#对第一显著的差异peaks画小提琴图pt.size = 0.1,idents = c("CD4 Naive","CD14 Mono")
)
plot2 <- FeaturePlot(object = pbmc,features = rownames(da_peaks)[1],#对第一显著的peaks画feature图pt.size = 0.1
)plot1 | plot2
除了使用DA test的方法,我们还可以通过比较两组细胞的Fold Change accessibility 来鉴定DA区域
这个方法可能比运行DA test的方法快很多,但这个方法没有做统计检验,所以无法避免一些潜在因素的影响,如细胞间的测序深度差异造成的影响
但使用这个方法可以快速对数据进行探索,FoldChange()函数可以进行上述操作
# 通过FoldChange函数比较两组细胞的Fold Change accessibility 来鉴定DA(differentially accessible)区域
fc <- FoldChange(pbmc, ident.1 = "CD4 Naive", ident.2 = "CD14 Mono")
head(fc)
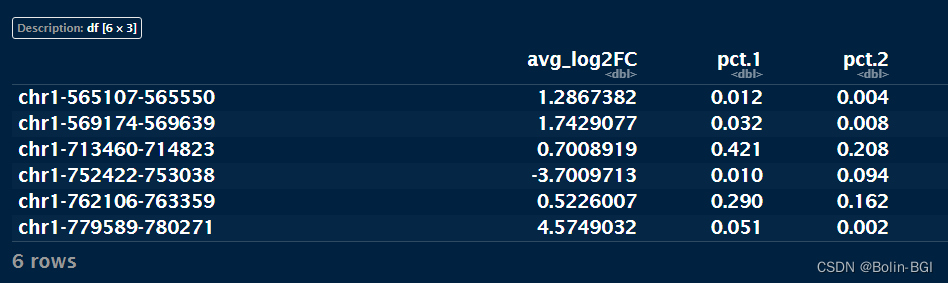
单纯的peak坐标很难单独进行解释,我们使用ClosestFeature()函数查找这些peaks的最近的基因。如果我们查看基因列表,会发现naive T细胞中的开放峰与BCL11B和GATA3(T细胞分化的关键调控因子)等基因接近,而单核细胞中开放的峰与CEBPB(单核细胞分化的关键调控因子)基因接近
在发现这些gene之后,还可以进行gene的富集分析,可以使用 GOstats 进行富集分析
open_cd4naive <- rownames(da_peaks[da_peaks$avg_log2FC > 3, ])
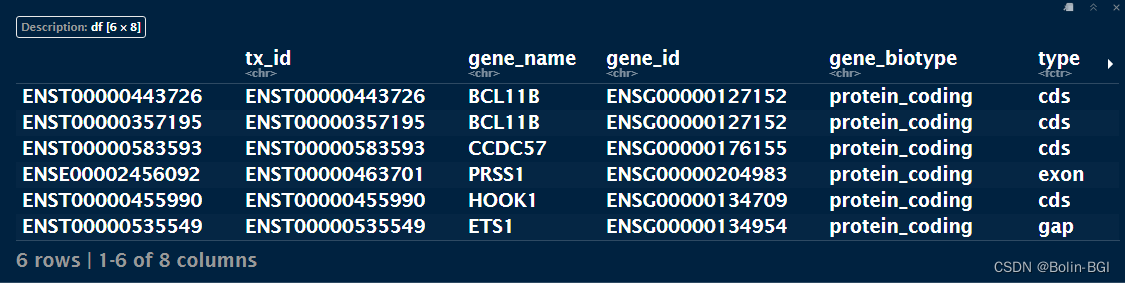
open_cd14mono <- rownames(da_peaks[da_peaks$avg_log2FC < -3, ])closest_genes_cd4naive <- ClosestFeature(pbmc, regions = open_cd4naive)
closest_genes_cd14mono <- ClosestFeature(pbmc, regions = open_cd14mono)head(closest_genes_cd4naive)head(closest_genes_cd14mono)
九、Plotting genomic regions(基因组ATAC信号可视化)
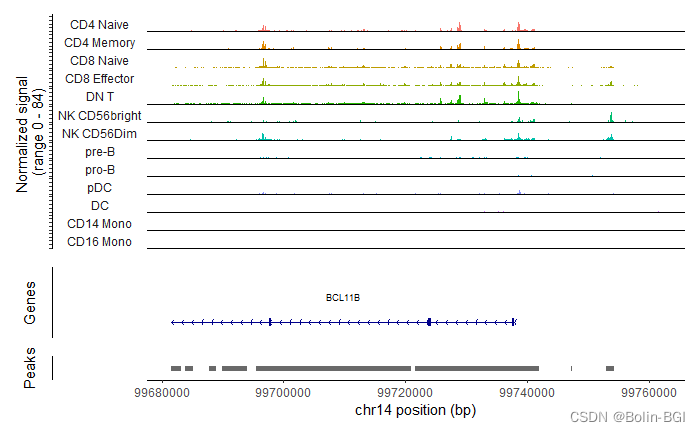
# 我们可以使用该函数绘制基因组区域间Tn5整合的频率,这些区域是根据细胞簇、细胞类型或存储在对象中的任何其他元数据进行分组的;
levels(pbmc) <- c("CD4 Naive","CD4 Memory","CD8 Naive","CD8 Effector","DN T","NK CD56bright","NK CD56Dim","pre-B",'pro-B',"pDC","DC","CD14 Mono",'CD16 Mono') CoveragePlot(object = pbmc,region = rownames(da_peaks)[1],extend.upstream = 40000,extend.downstream = 20000
)
参考文献:
Signic官方:https://stuartlab.org/signac/articles/pbmc_vignette.html
简书:https://www.jianshu.com/p/80ce771bb921
这篇关于scATAC-seq(Signic)官方分析流程的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!



