本文主要是介绍Microbiome | 山东大学与马普所揭示不同大型藻类共有附生细菌类群,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
揭示不同大型藻类共生菌群的核心细菌
Epiphytic common core bacteria in the microbiomes of co-located green (Ulva), brown (Saccharina) and red (Grateloupia, Gelidium) macroalgae

- 导读 -
2023年6月,山东大学海洋学院、微生物技术国家重点实验室杜宗军教授课题组和马克斯普朗克研究所海洋微生物研究所的Hanno Teeling博士课题组在Microbiome期刊合作发表了题为“Epiphytic common core bacteria in the microbiomes of co-located green (Ulva), brown (Saccharina) and red (Grateloupia, Gelidium) macroalgae”的研究论文,对同一采样点中不同大型藻类共有附生细菌类群开展了深入研究。联合培养博士生卢德臣为论文的第一作者,马克斯普朗克研究所海洋微生物研究所的博士生王凤青以及Rudolf I Amann教授为共同作者。
- 摘要 -
本研究从中国威海沿海一个固定的礁石上对石莼(Ulva sp.,绿藻)、海带(Saccharina sp.,褐藻)、石花菜(Grateloupia sp.,红藻)和蜈蚣藻(Gelidium sp.,红藻)以及海水和沉积物进行采样,使用16S rRNA扩增子测序,对样品中的细菌域微生物进行了鉴定,共鉴定了14个核心属(在所有大型藻类中都属于核心类群)和14个优势属(至少在三种大型藻类中属于核心类群)。藻际核心属占所有属类群的0.7%,但平均占细菌总丰度的51.1%。对这些样品中的细菌进行了培养分离,总共分离到可培养细菌5,527个菌株(其中大型藻际细菌4,426个),代表1,235个物种(其中685个潜在新物种)。对选定的菌株进行了基因组测序,得到820个非冗余基因组草图(506个潜在新物种)。对23个样品进行了宏基因组测序,得到1,619个宏基因组组装基因组(MAG),代表另外1,183个非冗余基因组。从28个核心/优势属中获得了230个分离株和153个基因组。分析了藻圈细菌降解藻类多糖和产生生物活性次级代谢产物的潜力,总共获得4,451个多糖利用位点(PUL)和8,810个生物合成基因簇(BGC)。多糖利用位点与次级代谢产物基因簇在核心/优势属中尤其丰富。通过对MAG和基因组的代谢注释和分析,为新的藻圈细菌物种及其生态位提供了新的见解,从而加深了对大型藻藻际微生物组的了解。
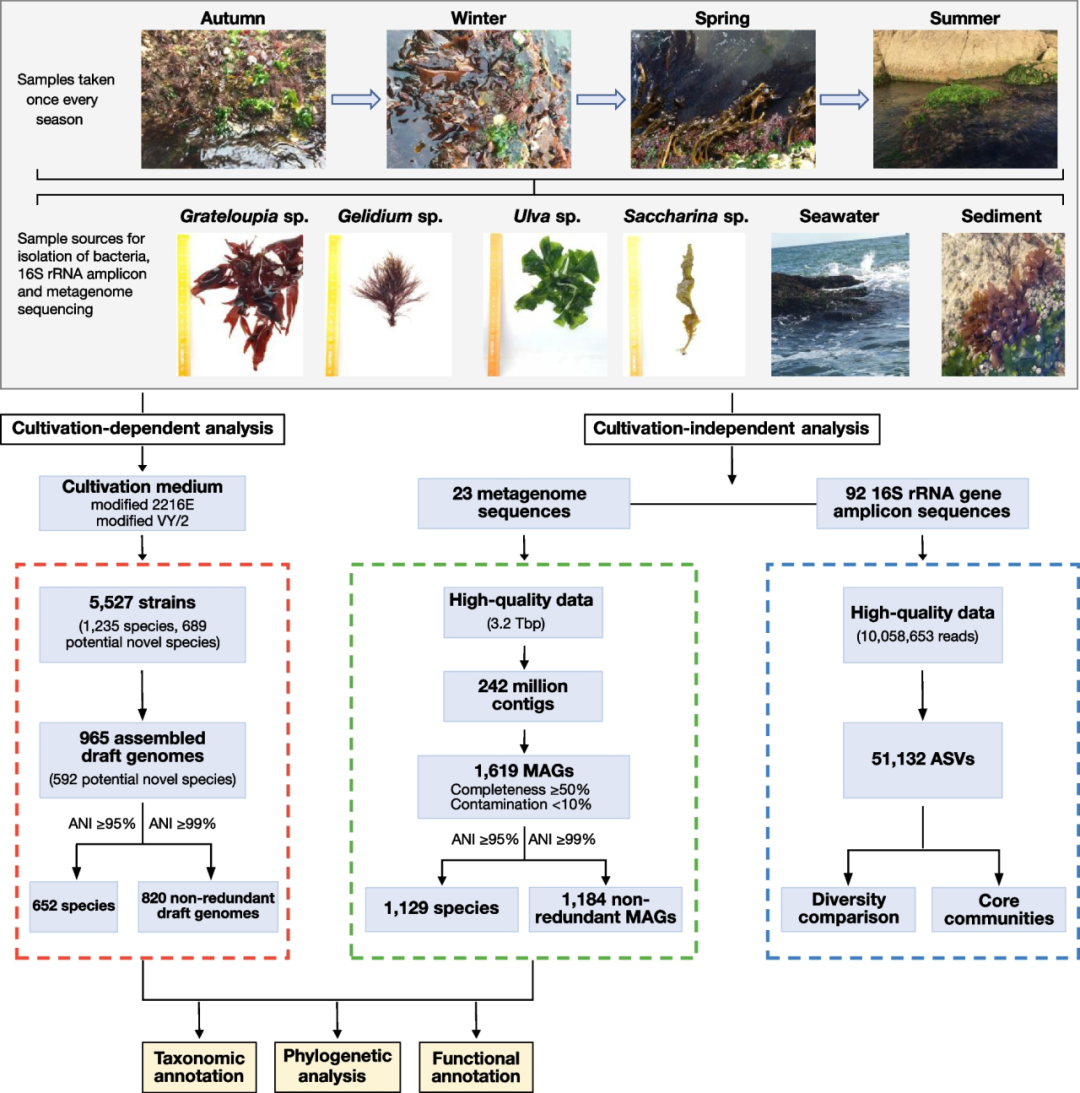
研究流程
- 研究背景 -
“大型藻类”涵盖三个主要谱系:红藻门、绿藻门和褐藻门,包含大约12,000个物种,在全世界沿海海洋生态系统中广泛分布。大型藻类表面有大量细菌定植,与大型藻类相关的细菌已与大型藻类共同进化了大约16亿年,并具有复杂而密切的关系。Bell和Mitchell在1972年将藻类与细菌密切相互作用的区域被称为“藻际”。藻际微生物组在组成和功能方面与周围海水中的微生物明显不同。藻际微生物通过提供生长因子支持大型藻类宿主实现形态发育、适应环境变化、藻类孢子的释放和沉降等。藻际微生物还含有潜在的有害细菌,例如病原体可以降解大型藻类组织的共生细菌。动态环境变化会影响它们的藻际群落组成,宿主形态也发挥着一定作用。尽管存在这种非生物影响,但各种研究表明藻际群落也具有宿主特异性。大型藻类藻际微生物的主要类群在微生物群落组成中占主导地位,对于维持藻际微生物的正常功能具有关键的调节作用。然而,对于不同大型藻类中常见的核心细菌在分类学、代表性基因组和生态生理功能方面缺乏研究。
大型海藻的生态作用与陆地植物相似,然而人们对这些藻际细菌的了解远少于对陆生植物(尤其是根际植物)相关细菌的了解。近年来,人们对海洋植物和藻类的藻际细菌的兴趣日益浓厚,超出了对微生物群落组成的单纯描述,例如最近对海藻和海带微生物组的研究。除了对维持定植模式机制的研究,藻际附生微生物的潜在的遗传功能及其工业应用的潜力也有待进一步探究。
- 结果 -
1. 所有藻类都具有相似但多样化的藻际群落,非核心类群具有显著的季节性
在ASV(amplicon sequence variants)的α多样性(丰富度)分析中,藻际样品表现出与海水相似的总体多样性,但多样性低于沉积物样品。使用Bray-Curtis相异性指数对ASV的β多样性进行主坐标分析(PCoA),显示按栖息地进行聚类,藻际菌群与沉积物和海水对照明显分开。然而,藻际样本的成对比较并没有发现显著差异,这表明采样的大型藻类物种之间存在相当程度的共享分类群。去除核心类群ASV后(即所有大型藻类上出现的类群),样本根据季节更加清晰地聚类,表明非核心类群对季节变化的贡献更大。UniFrac UPGMA聚类分析证实沉积物、海水和藻际栖息地之间存在显著差异。与海水样品相比,藻际样品中Bacteroidota的相对丰度通常较高。仅在春季,Bacteroidota的丰度高达25.1%。大多数季节来自同一大型藻类的样本都聚集在一起,特别是春季的Ulva sp.、Grateloupia sp.和Gelidium sp.,表明藻际群落特别相似。
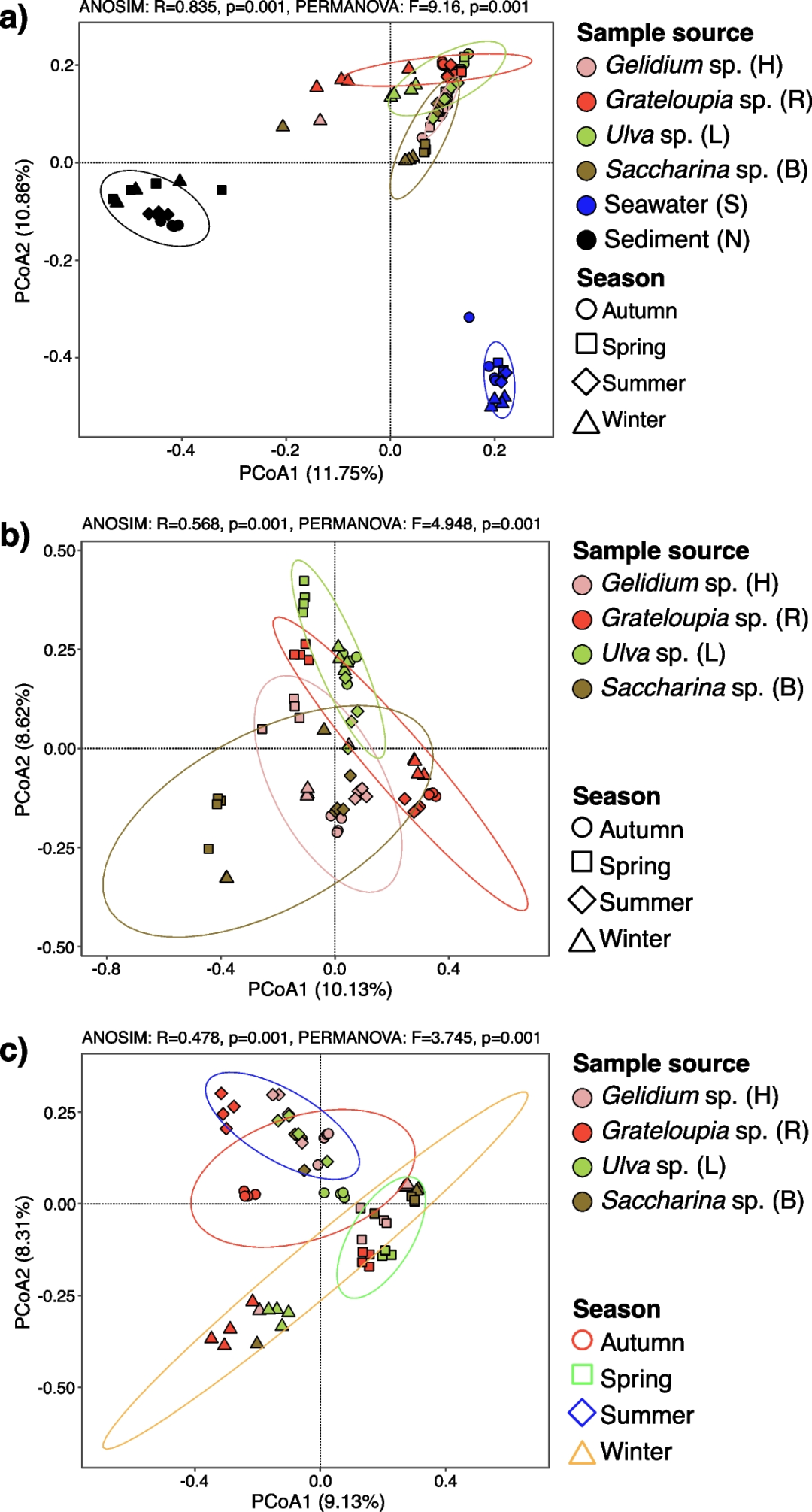
图1 使用未加权 UniFrac 距离计算的样本和季节的 Bray-Curtis 相似性的主坐标分析 (PCoA) 图(每个点对应于单个样本)
2. 藻际微生物以少数稳定的核心藻际类群为主
ASV分析显示,藻际中的大多数细菌科仅由一两个属代表,而Flavobacteriaceae和Rhodobacteraceae等少数细菌则具有更广泛的代表性。这种较低的整体均匀度强调了藻际群落主要由少数丰富的进化枝主导。来自8个科(Proteobacteria、Bacteroidota、Verrucomicrobiota和Actinobacteriota)的14个核心属存在于所有大型藻类中,至少在一个样品中丰度超过1%。核心藻际属平均占所有藻际属的1.4%(Gelidium sp.,14/972、Grateloupia sp.,14/1,000、Ulva sp.,14/973和Saccharina sp.,14/870),但平均占所有藻际细菌相对丰度的43.5%(Gelidium sp.)、53.9%(Grateloupia sp.)、58.3%(Ulva sp.)和48.8%(Saccharina sp.)。相比之下,海水和沉积物样品中这些核心藻际属的平均相对丰度分别仅为5.7%和1.5%。四种大型藻类中的三种中大量存在另外十四个属,以下称为藻际优势属。所有藻类中14个核心属及14个优势属的在季节中的相对丰度的变化具有类似的模式。
尽管栖息地之间(大藻、海水与底泥)的差异在科和属水平上更加明显,但大型藻类藻际之间仍具有很高的一致性。在四种所有研究的藻类物种中的属和科水平上观察到稳定的核心藻际组成,特别是对于Alphaproteobacteria、Gammaproteobacteria和Bacteroidota的主要成员。核心属虽然只代表藻际多样性的一小部分,尽管它们的相对比例在整个季节波动,但构成了藻际丰度的主要部分。例如,Granulosicoccus已被证明栖息在多种大型藻类物种上。采样的礁石中发现一些常见且广泛存在的潜在藻际细菌,其中一些细菌,特别是核心/优势属的成员,在大型藻类定植方面比其他细菌更成功。
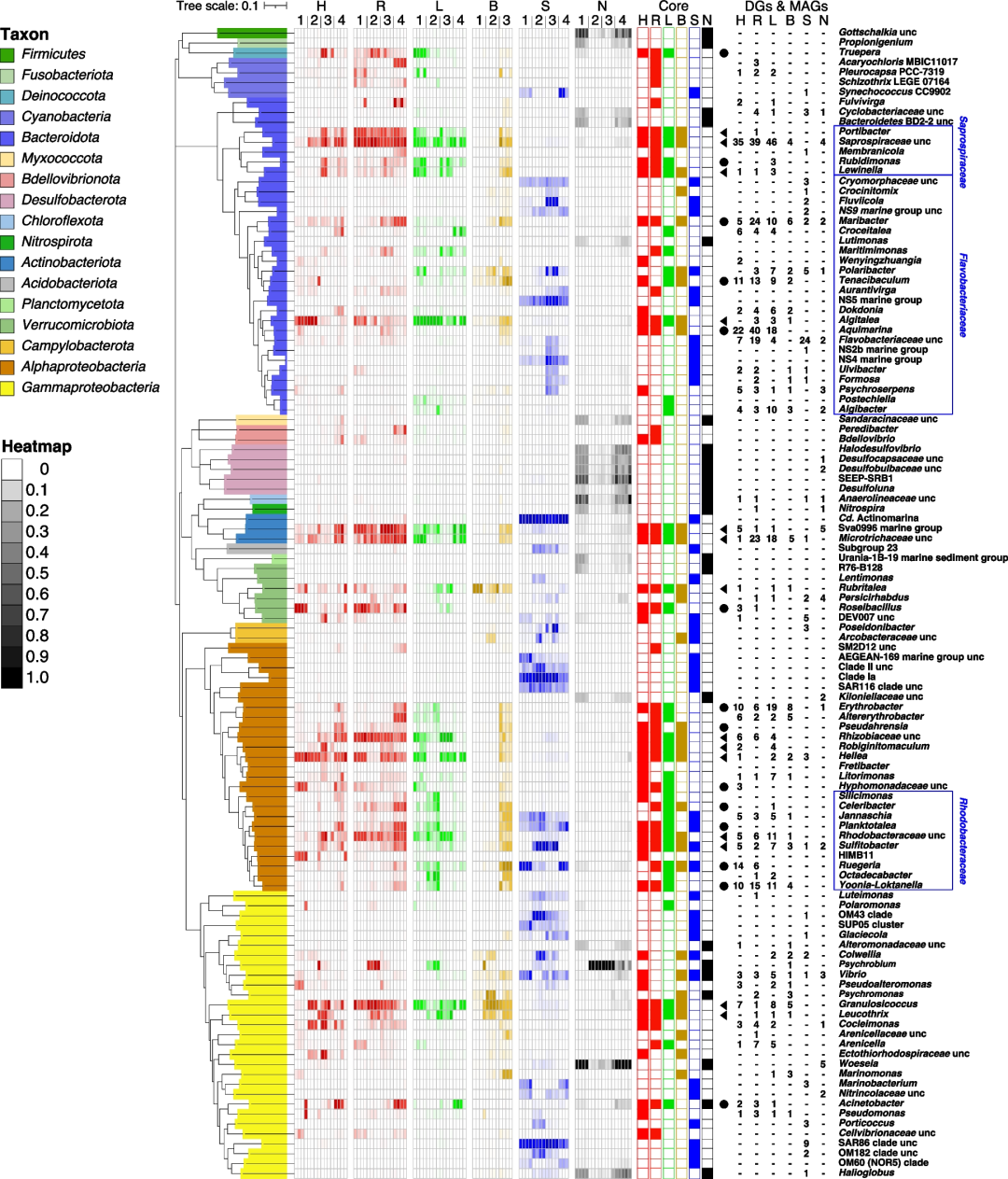
图2 116 个核心属的系统发育分析、相对丰度及其基因组数量信息
3. 来自藻际细菌(包括新物种)的大量基因组草图和MAG扩大了已知藻际细菌种类的目录
大多数来自海洋大型藻类的细菌都抵抗普通的培养技术,而那些已培养的细菌大多属于“稀有生物圈”。在这项研究中,共培养到367个属(大型藻类:302)的菌株,其中包括6个属(Hellea、Algitalea、Sulfitobacter、Granulosicoccus、Leucothrix、Robiginitomaculum)核心菌株和10个属(Maribacter、Tenacibaculum、Aquimarina、Erythrobacter、Planktotalea、Yoonia-Loktanella、Ruegeria、Acinetobacter、Pseudahrensia、Celeribacter)优势藻际属。
基于16S rRNA序列相似性,选择了965个(大型藻类:864)菌株进行基因组草图测序,其中包括550个冗余新种和42个冗余新属,补充了尚未有参考基因的大量物种。从宏基因组中,共获得了1,619个(大型藻类:936)MAG,包含1,184个非冗余MAG(99% ANI,即average nucleotide identity)代表的1,129个物种(95% ANI)。总共961个基因组草图和545个MAG的完整性>90%,污染估计<5%。与公共数据库工具GTDB-Tk比较后,82.7%(795/961)的基因组草图和88.4%(482/545)的高质量MAG不属于任何描述的物种。为了确定物种总数,还使用基于multi-step distance-based的方法(95% ANI)对最初的965个基因组草图和1,619个MAG进行了聚类。结果产生了1,781种(大型藻类:1,185)推断的原核物种、1,689种细菌(大型藻类:1,182种)和49种古生菌(大型藻类:3)。
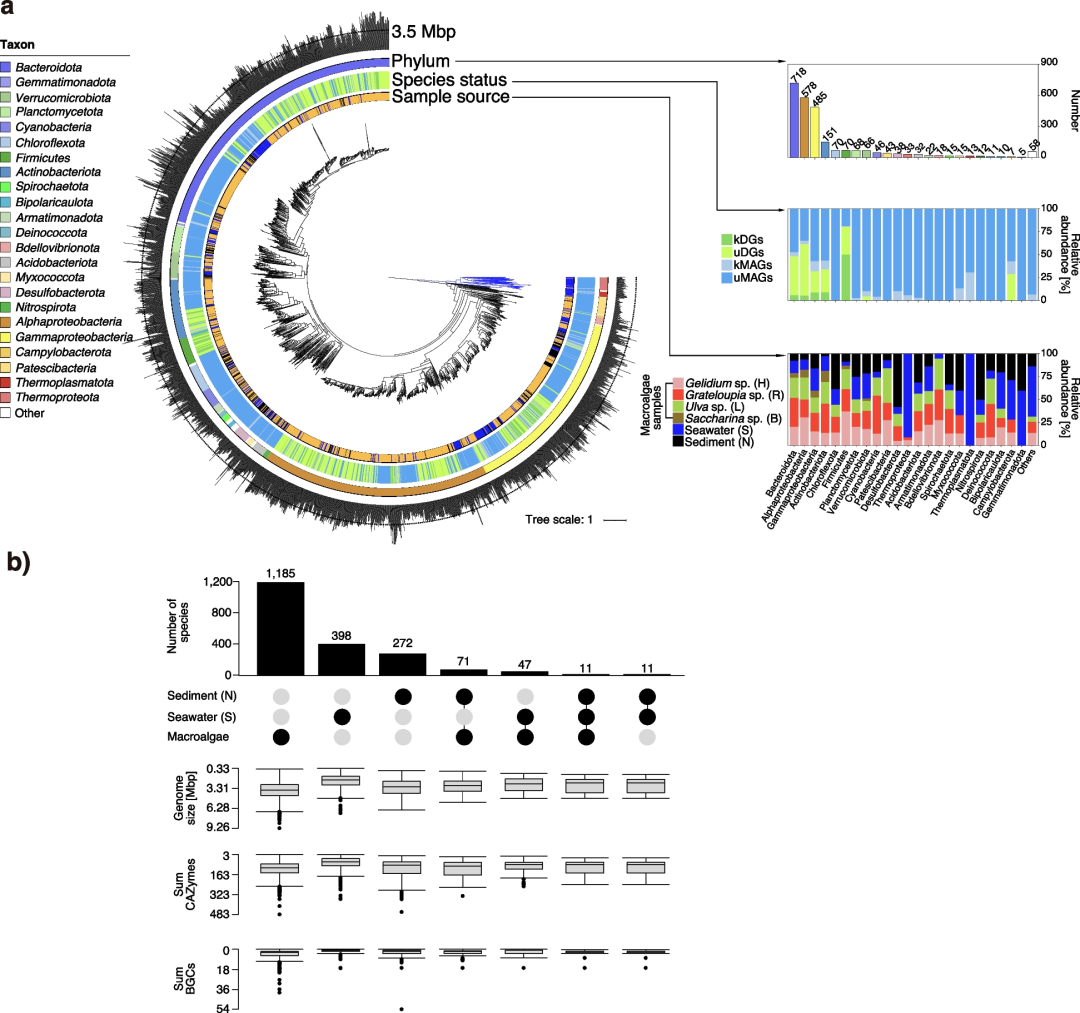
图3 宏基因组组装的基因组和基因组草图的分类、样品来源及数量统计
4. 藻际拟杆菌门含有高比例的未知基因并主导了多糖的降解
基于EggNOG v5、COG (2020)、Pfam (2020)、UniProtKB和KEGG数据库进行自动注释后,结果显示从培养的拟杆菌门获得的376个基因组(305个来自大型藻类)的未知基因比例最高。这说明大型藻类定植的Bacteroidota具有特别丰富的未知功能的基因资源。此外,来自大藻藻际细菌的基因组平均大于来自沉积物和海水细菌的基因组,其中海水样品的平均基因组最小。作者搜索了所有基因组草图和MAG中的碳水化合物活性酶(CAZyme)基因,并鉴定了292,848个同源物。Bacteroidota(717)、Chloroflexi(70)、Planctomycetota(68)、Verrucomicrobiota(66)、Acidobacteriota(32)和Actinobacteriota(151)编码最高比例的分解代谢CAZymes,即糖苷水解酶(GH)、碳水化合物酯酶(CE)、碳水化合物结合模块(CBM)、辅助活性酶(AA)和多糖裂解酶(PL)。大多数(61.8%)CAZyme基因在Bacteroidota中发现,证实了该门成员在藻类多糖降解中发挥的关键作用。预测的CAZymes包括30.6% GH、29.9% GT、15.1% CE、10.2% CBM、5.1% PL和5.1% AA,并且这些比例在样品中相似。GT(2.4%)和AA(1.7%)的信号肽预测很少,而PL(76.5%)、GH(55.6%)和CE(42.9%)的信号肽预测比例要高得多,表明其周质或细胞外定位。硫酸化藻多糖脱硫所需的预测分泌硫酸酯酶的比例在海水和沉积物中比在藻际细菌中高约11%和约13%,Planctomycetota和Verrucomicrobiota具有大量CAZyme和硫酸酯酶基因。
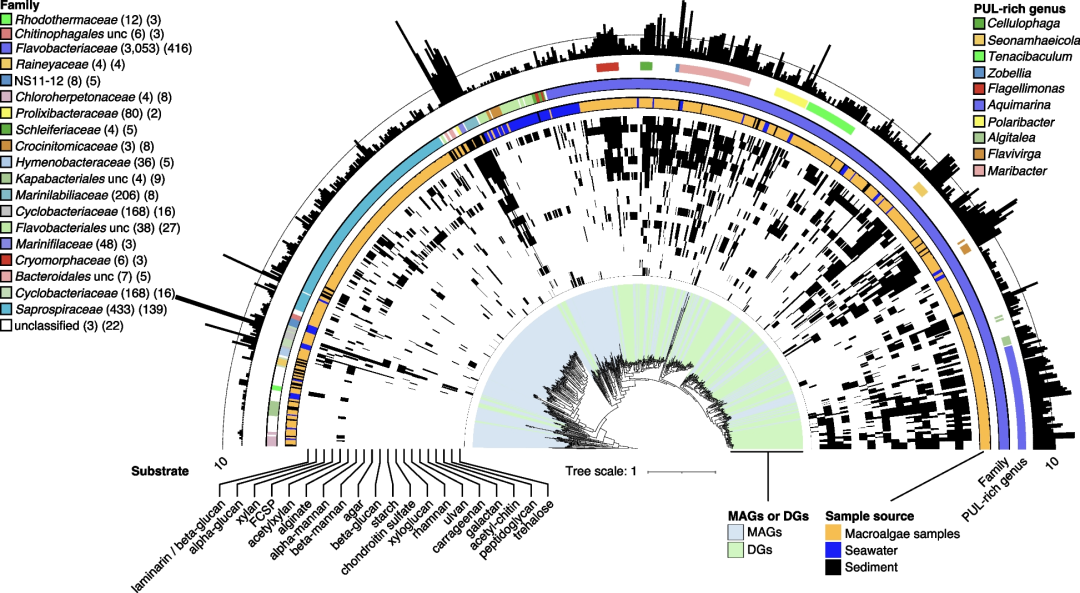
图4 MAG 和基因组草案 (DG) 中的 PUL 分布及底物统计分析
5. PUL的多糖水解酶复杂度及其底物间的关系
大型藻类多糖化学结构的变化不仅取决于物种,还取决于大型藻类样本的个体部位和发育阶段、季节和其他环境因素。单个PUL通常编码整个装置以降解特定聚糖,但对具有复杂化学结构的聚糖的降解,已有研究表明可能涉及多个PUL的共同作用。这可能解释了为什么在Bacteroidota中作者不仅观察到大量的PUL,而且还观察到CAZyme基因的高度多样性,特别是在大型融合PUL中。藻际Bacteroidota的PUL基因库和多样性表明高度的功能冗余,这可能能够适应各种大型藻类宿主。这种冗余可能是通过水平基因转移获得PUL的结果。后者的表现是PUL模式也不总是与16S系统发育一致。特别是所有采样栖息地的拟杆菌门都富含PUL,这支撑了Bacteroidota在海洋多糖降解中发挥的特殊作用,其成员可以在功能上相互补充。
作者预测出4,451个PUL、6,376个PUL-like基因簇、19,826个CGC和1,699个susCD基因对。大多数发现于纯培养基因组中(3,461、3,875、9,572和1,076)。由于整体完整性更高,在纯培养基因组中发现了大约3.5倍的PUL。与海水相比,藻际中的PUL数量高出1.6倍,沉积物基因组中的PUL数量高出2.8倍。在藻际中,富含PUL的物种主要属于Zobellia、Polaribacter、Aquimarina、Tenacibaculum、Algitalea和Maribacter,代表了藻际的核心属或优势属。其他富含PUL的属包括Cellulophaga、Flagellimonas、Flavavivirga和Seonamhaeicola。最常见的预测底物是预测的含木糖的多糖(779)、β-葡聚糖/昆布多糖(618)、α-葡聚糖(482)、含岩藻糖的硫酸化多糖(FCSP)(444)、藻酸盐(426)、α-甘露聚糖(268)、β-甘露聚糖(220)、硫酸化含α-鼠李糖的多糖(219)、琼脂(192)、软骨素(158)、木葡聚糖(133)半乳聚糖(128)、绿藻硫化多糖(127)、淀粉(114)、角叉菜胶(109)、几丁质(109)、果胶(72)、肽聚糖(69)、果聚糖(36)和卟啉(31)。预测靶向结构明确的简单多糖(如昆布多糖、淀粉和藻酸盐)的PUL包含较少的CAZyme基因,并且比预测靶向更复杂的多糖(如卡拉胶和绿藻硫化多糖)的PUL更保守。一些较大、复杂的PUL实际上可能会利用多种多糖底物。例如,SusC/D蛋白质树中的簇27_1包含具有GH5_2基因的角叉菜胶PUL。后者可能针对木聚糖(内切-β-1,4-木聚糖酶功能)或纤维素(内切-β-1,4-葡聚糖酶功能),它们通常与自然栖息地中的角叉菜胶共存。同样,预计靶向绿藻硫化多糖和鼠李糖半乳糖醛酸的PUL含有看似与实际底物无关的额外内切水解酶。原因可能是藻类硫酸化多糖很少是同质的,而大多是不同聚糖的复杂异质混合物。
6. 藻际类群,特别是藻际核心类群具有丰富的生物合成基因簇
作者确定了8,810个假定的BGCs。预测的产物类别包括萜烯(28.3%)、细菌素(12.3%)、非核糖体肽(NRPS)(10.5%)和NRPS样簇(8.0%)、高丝氨酸内酯(7.8%)、III型聚酮合酶(7.5%)、I型聚酮合酶(5.9%)和β-内酯(5.4%)。由于基因组草图通常比MAG更完整,因此它们的不完整BGC比例较低。基因组草图中预测的4,816个BGC中有20.1%位于Contig边缘,因此可能不完整,而MAG中预测的3,994个BGC中有73.2%属于这种情况。作者观察到门之间存在明显的区别,但在栖息地方面没有观察到BGC家族的明显趋势。最大BGC基因簇是从石花菜属中培养的链霉菌属物种,它编码不少于22个PKS和NRPS模块。BGC数量最多的前100个基因组中有93个属于藻际细菌,BGC数量最多的前20个基因组中有10个属于藻际拟杆菌门。后者表明该门产生次生代谢产物的潜力可能仍被低估。拟杆菌门具有高比例的用于萜烯和NRPS生物合成的BGC,例如新的核心藻际物种Aquimarina sp. 2-328。
大多数BGC在Bacteroidota、Alphaproteobacteria、Gammaproteobacteria、Firmicutes和Actinobacteriota中被鉴定,所有类群都富含核心藻际细菌。核心/优势藻际类群,例如Flavobacteriaceae和Rhodobacteraceae的Maribacter、Algitalea、Tenacibaculum、Aquimarina、Ruegeria和Sulfitobacter属的成员包含具有非常高的BGC比例。Firmicutes和Actinobacteriota因产生丰富的次生代谢产物而闻名。作者在151个Actinobacteriota基因组(包括100个MAG)中发现了559个BGC,涵盖了预测产物的广泛多样性。虽然在沉积物中Firmicutes的MAG中发现了最高数量的BGC(54),但从大型藻类中分离的Streptomyces菌株3-371的基因组中发现了第二高数量(39)和第三高数量的BGC(36)。Alphaproteobacteria尤其富含BGC,许多编码高丝氨酸内酯,特别是核心藻际科Rhodobacteraceae,其在控制藻类表面定植的微生物组成过程中发挥关键作用。例如,从Gelidium sp.分离的物种Roseovarius sp. 3-342(Rhodobacteraceae)包含6个相关基因簇。大多数预测的BGC产物都是未分类的,这反映了藻际细菌具有尚未探索的生物合成功能的丰富资源。
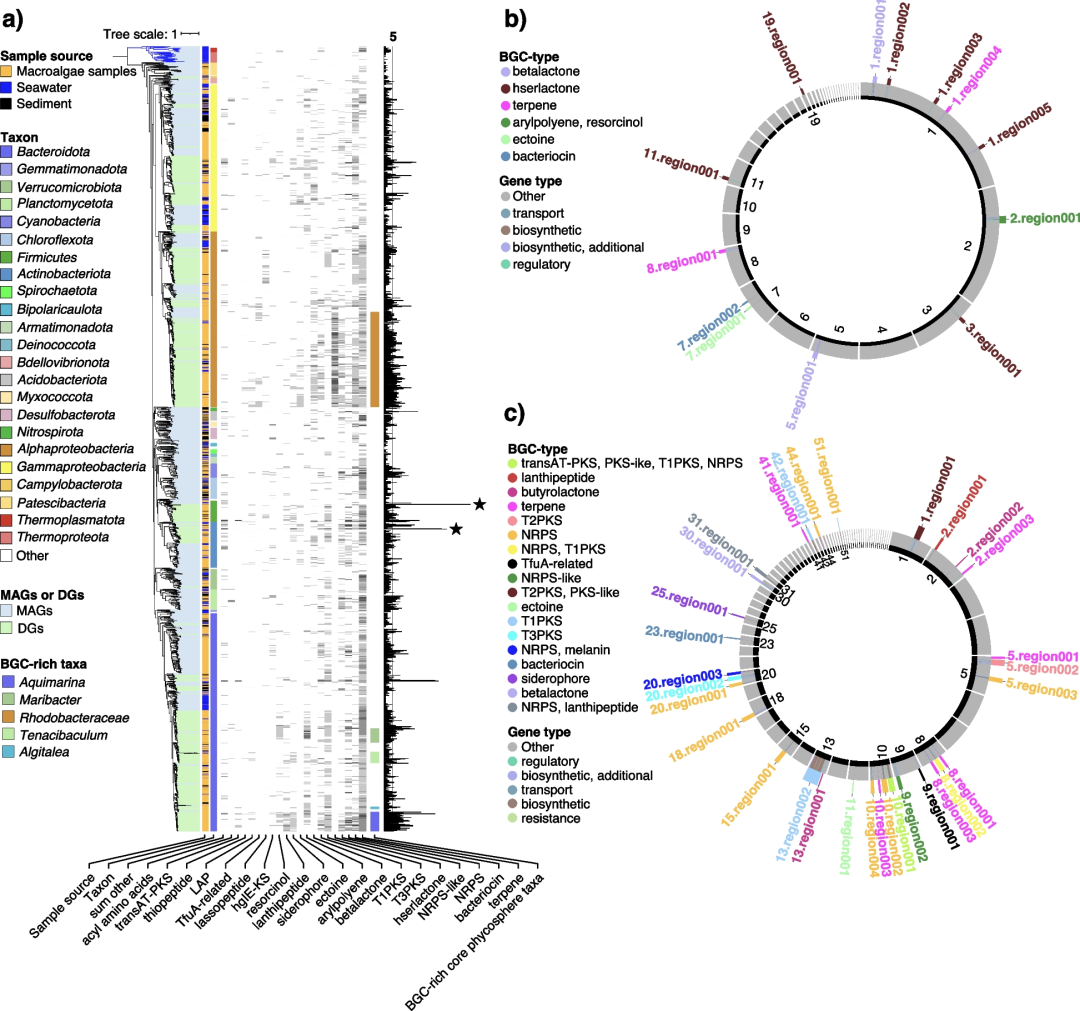
图5 1,619 个 MAG 和 965 个基因组草图 (DG) 中的生物合成基因簇组成和分布
- 文章结论与展望 -
综上所述,本研究综合利用依赖于培养以及免培养的宏基因组的方法、采用生物信息学分析方法等研究手段,确定了在科和属水平上与四种大型海藻相关的核心细菌群落组成,扩展了藻际微生物的组装模式。本研究仅探究了一个采样点的四种大型藻类,这对于在全球尺度的更多样的藻类物种的大藻藻际微生物群落组装特点及功能研究具有一定的借鉴意义。本研究最重要的工作是对956株不同样品来源的菌株(568个候选新物种)进行了基因组测序,通过对23个宏基因组(共1.3T的原始数据)的分析,得到了1,619个中高质量的MAGs。这些基因组极大的提高了藻际微环境中的代表性基因组的覆盖度,提供了目前为止关于大藻藻际微生物最大的参考基因组目录(包括可培养类群的基因组)。本研究发现藻际核心类群相对于其他环境具有更大的基因组,更多的多糖降解潜力以及更多的次级代谢产物合成潜力,为未来挖掘多糖降解酶及次级代谢产物提供了新的菌种资源。对细菌的PULs和BGCs的多样性进行研究发现,在Bacteroidota中总共获得4,000多个PUL,这些PUL对应着多种多样的多糖底物。在2,548个基因组中注释到8,000多个BGCs,多种多样的BGCs为后续的海洋来源的次级代谢产物的挖掘研究提供了丰富的候选材料。本研究提供的基因组数据将为未来整个大藻微生物组的宏转录组研究提供有价值的搜索空间。迄今为止,部分丰富的异养藻际细菌,特别是浮霉菌门、疣微菌门和绿弯菌门等类群仍未成功大规模培养,这将成为未来研究的重点。
- 参考文献 -
Lu, DC., Wang, FQ., Amann, R.I. et al. Epiphytic common core bacteria in the microbiomes of co-located green (Ulva), brown (Saccharina) and red (Grateloupia, Gelidium) macroalgae. Microbiome 11, 126 (2023). https://doi.org/10.1186/s40168-023-01559-1
- 作者介绍 -

山东大学
卢德臣
博士后
2015级山东大学与马克思普朗克研究所海洋微生物研究所联合培养博士生,微生物学,理学博士,E-mail:DechenLu@hotmail.com。
研究方向:
1、整合多数据库的生物信息学工具开发;
2、微生物功能基因簇的进化与功能模式的相关研究;
3、生物大分子的微生物降解及生物能源转化研究。

马克斯普朗克海洋微生物研究所
王凤青
博士研究生
研究方向:
1、基于16S rRNA 扩增子的微生物群落组成及多样性分析;
2、使用生物信息学的方法分析基于PacBio或Illumina测序平台的基因组、宏基因组、宏蛋白组以及宏转录组等序列;
3、浮游植物-浮游细菌的相互作用;
4、海洋细菌的多糖代谢过程分析;
5、海洋碳循环。

马克斯普朗克海洋微生物研究所
Rudolf Amann
教授
Rudolf Amann教授主要从事微生物生态学、海洋碳循环、浮游植物-浮游细菌的相互作用、微生物分类学、通过FISH进行单细胞鉴定等方面的研究,他还担任了马克斯普朗克海洋微生物研究所分子生态学部门主任。

马克斯普朗克海洋微生物研究所
Hanno Teeling
博士,学科带头人
目前的研究兴趣是综合宏基因组数据分析的生物信息学方法,浮游植物与细菌的相互关系,重点是碳的周转,以及极端栖息地的微生物群落。
其他研究兴趣:
1、海洋植物菌的细胞生物学和生态生理学作用;
2、海洋黄杆菌的有机物降解作用;
3、嗜盐细菌和古细菌;
4、下一代测序方法和后续数据分析。

山东大学
海洋学院
杜宗军
教授、博士生导师、副院长、学术委员会主任委员
建立了山东省海洋微生物菌种保藏与应用工程技术研究中心,保藏微生物1.5万余株,平台提供菌种鉴定、共享、筛选等技术服务(http://sdum.wh.sdu.edu.cn);致力于细菌分离培养技术以及分类学研究,在细菌分离培养方面形成了独特的技术体系。发现了大量具有重要研究价值的细菌新类群,例如海洋沉积物中细菌区系的核心成员——伍斯菌科。发表海洋细菌新纲1个(童第周菌纲),新目3个,新科5个,新属40余个。杜宗军教授还担任了山东省海洋微生物菌种保藏与应用工程技术研究中心主任、威海市海洋微生物技术重点实验室主任、山东大学海洋生物工程技术研究中心主任。
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文
这篇关于Microbiome | 山东大学与马普所揭示不同大型藻类共有附生细菌类群的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!




