本文主要是介绍蛋白结构预测模型评价指标,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
| 欢迎浏览我的CSND博客! Blockbuater_drug …点击进入 |
|---|
前言
本文汇总了AlphaFold和AlphaFold-multimer等蛋白结构推理预测中,不同蛋白结构预测模型的评价指标。
一、蛋白结构预测模型评价指标
TM-score
TM-score是一种用于评价蛋白质结构拓扑相似性的度量。由张yang首先提出,该方法解决了传统度量方法如均方根偏差(RMSD)中存在的两个主要问题:(1)TM评分对较小距离误差的权重大于对较大距离误差的权重,使得评分值对全局折叠相似性的敏感性大于对局部结构变化的敏感性;(2)TM-score引入了长度相关的尺度对距离误差进行归一化,并使得TM-score的大小与随机结构对的长度无关。计算公式如下:

该作者实验室提供了计算TM-score的C++程序,可编译后使用,方法如下:
wget https://zhanggroup.org/TM-score/TMscore.cppg++ -static -O3 -ffast-math -lm -o TMscore TMscore.cpp
## Run TM-score to compare 'model' and 'native':
TMscore model native
## Run TM-score to compare two complex structures with multiple chains
## Compare all chains with the same chain identifier
TMscore -c model native
将RCSB pdb结构与AlphaFold2预测结果对比,结果如下:
./TMscore ranked_0.pdb 8i55.pdb ************************************************************************** TM-SCORE ** A scoring function to assess the similarity of protein structures ** Based on statistics: ** 0.0 < TM-score < 0.17, random structural similarity ** 0.5 < TM-score < 1.00, in about the same fold ** Reference: Yang Zhang and Jeffrey Skolnick, Proteins 2004 57: 702-710 ** For comments, please email to: zhanglab@zhanggroup.org **************************************************************************Structure1: ranked_0.pdb Length= 143
Structure2: 8i55.pdb Length= 120 (by which all scores are normalized)
Number of residues in common= 120
RMSD of the common residues= 0.581TM-score = 0.9811 (d0= 4.05)
MaxSub-score= 0.9753 (d0= 3.50)
GDT-TS-score= 0.9917 %(d<1)=0.9667 %(d<2)=1.0000 %(d<4)=1.0000 %(d<8)=1.0000
GDT-HA-score= 0.9271 %(d<0.5)=0.7417 %(d<1)=0.9667 %(d<2)=1.0000 %(d<4)=1.0000-------- rotation matrix to rotate Chain-1 to Chain-2 ------i t(i) u(i,1) u(i,2) u(i,3)1 -16.8570056544 0.9777658128 -0.0973231336 0.18574774052 17.8632659480 -0.0324689163 0.8048346382 0.59261030593 11.0145890770 -0.2071709074 -0.5854651253 0.7837798174Superposition in the TM-score: Length(d<5.0)= 120
(":" denotes the residue pairs of distance < 5.0 Angstrom)
MEALVLVGHGSRLPYSKELLVKLAEKVKERNLFPIVEIGLMEFSEPTIPQAVKKAIEQGAKRIIVVPVFLAHGIHTTRDIPRLLGLIEDNHEHHHEHSHHHHHHHHHEHEKLEIPEDVEIIYREPIGADDRIVDIIIDRAFGR
::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::: :::::::::::::::::::::::::::::::
MEALVLVGHGSRLPYSKELLVKLAEKVKERNLFPIVEIGLMEFSEPTIPQAVKKAIEQGAKRIIVVPVFLAHGIHTTRDIPRLLGLIED-----------------------EIPEDVEIIYREPIGADDRIVDIIIDRAFGR
12345678901234567890123456789012345678901234567890123456789012345678901234567890123456789012345678901234567890123456789012345678901234567890123lDDT
局部距离差异测试(Local Distance Difference Test,lDDT)是一种无叠加的评分,用于评估模型中所有原子的局部距离差异,包括立体化学变换的验证。参考物可以是单个结构,也可以是结构的集合。
蛋白质结构预测技术的评估需要客观的标准来衡量计算模型和实验确定的参考结构之间的相似性。传统的基于碳α原子全局叠加的相似性度量受到结构域运动的强烈影响,并且不能评估模型中局部原子细节的准确性。lDDT是非常适合评估本地模型的质量,即使在存在的结构域运动,同时保持良好的相关性。
LDDT计算方法一:
在线计算:SWISS-MODEL lDDT
LDDT计算方法二:
python版本:感谢CSDN博主的贡献,python版本的lDDT score的计算可参考博文:蛋白结构评价-LDDT score
二、Alphafold中的评价指标
pLDDT
AlphaFold 预测结构所有残基Cα原子的lDDT被称为pLDDT score(per-residue lDDT-Cα),范围在0-100,分数越高,置信度越高。
Deepmind提供的算法如下:  作为一种基于lDDT的置信度度量,pLDDT也反映了结构(structure)中的局部置信度,应该用来评估单个结构域内的置信度。基于语言模型的蛋白结构预测方法如ESMFold也使用基于plDDT的度量方法。
作为一种基于lDDT的置信度度量,pLDDT也反映了结构(structure)中的局部置信度,应该用来评估单个结构域内的置信度。基于语言模型的蛋白结构预测方法如ESMFold也使用基于plDDT的度量方法。
plDDT 存储于AlphaFold及ESMFold预测结构(mmCIF或PDB文件)B因子字段(B-factor fields)中,即文件中ATOM记录的第11列。
pLDDT≥90:残基具有非常高(very high)的模型置信度
90 > pLDDT ≥70:残基则被归为确信的(confident)
70 > pLDDT ≥50:残基置信度较低(low)
pLDDT < 50:残基置信度很低(very low)。
最近的研究表明,非常低的置信度pLDDT分数与蛋白结构固有无序(intrinsic disorder)的高倾向相关。
 采用不包含在训练集中新近报道的PDB数据集(该数据集仅限于报告分辨率<3.5Å的结构,包括的链条总数为10215条)。比较了数据基于解析区域的每个残基lDDT-Cα和每个残基pLDDT。二者有很好的相关性:lDDT-Cα = 0.997 × pLDDT − 1.17 (Pearson’s r = 0.76)
采用不包含在训练集中新近报道的PDB数据集(该数据集仅限于报告分辨率<3.5Å的结构,包括的链条总数为10215条)。比较了数据基于解析区域的每个残基lDDT-Cα和每个残基pLDDT。二者有很好的相关性:lDDT-Cα = 0.997 × pLDDT − 1.17 (Pearson’s r = 0.76)


pTM
pLDDT头预测了lDDT-Cα值,这是一个成对操作的局部误差度量,但根据设计,它对使用单个全局旋转和平移可以对齐的残基部分不敏感。这对于模型是否准确预测长链整体结构域堆叠是不利的,为此,作者使用了全局叠加度量TM得分的预测器 TM-score。
特别地,我们可以预测可能通过实验解析的残基,并使用它们来产生预测的TM分数(pTM),其中每个残基的贡献通过其被解析的概率进行加权。
加权的目的是降低预测的非结构化部分的权重,从而产生更好地反映模型对确实存在的结构域的度量。



在最新的不再训练集中的PDB数据集上,pTM与实际TM-score相关性良好。
TM-score = 0.98 × pTM + 0.07 (Pearson’s r = 0.85)

PAE
预测对齐误差( Predicted Aligned Error,PAE )是AlphaFold系统的另一个输出结果。AlphaFold DB提供给结构PAE的图片和数据.json文件。
它表示如果预测结构和实际结构在残基y (使用Cα、N和C原子)上对齐,显示在残基x处的期望位置误差。PAEs以A 为单位测定,上限为31.75A。可以利用这些数值来评估模型(例如两个结构域)不同部分的相对位置和朝向(orientation)的置信度。注意,PAE是不对称的;因此,(x,y)和(y,x)的PAE值之间可能存在差异。
对于两个不同结构域中的残基x和y,如果PAE值( x , y)较低,则AlphaFold预测结构域具有明确的(well-defined)相对位置和取向。如果PAE值很高,那么这两个结构域的相对位置和方向是不可靠的

三、AlphaFold-multimer 蛋白结构的评价指标
2021年10 月4 日,DeepMind 团队推出了AlphaFold-Multimer,用于蛋白质复合物的预测,尤其是对同源或者异源复合物,AlphaFold-Multimer 对接预测评分均有提升,弥补了AlphaFold2的不足。

DockQ
DockQ是一种用于评估蛋白质-蛋白质分子对接模型的工具和指标。它被广泛用于评估蛋白质分子对接方法的性能和预测模型的准确性。范围0-1之间,越大准确性越好。
DockQ是三个指标的综合评分,即Fnat、LRMS和iRMS,
 Fnat: 预测复合体在交界面上的作用残基在真实复合体中的比例。
Fnat: 预测复合体在交界面上的作用残基在真实复合体中的比例。
LRMS: 把预测的复合体和真实复合体的两条链中比较长的链比对,短链的RMSD。
iRMS: 度量界面上两个原子相距10Å内的原子的集合的RMSD。
DockQ的计算方法:
https://github.com/bjornwallner/DockQ/
度量复合体预测准确性的指标:
***********************************************************
* DockQ *
* Scoring function for protein-protein docking models *
* Statistics on CAPRI data: *
* 0 < DockQ < 0.23 - Incorrect *
* 0.23 <= DockQ < 0.49 - Acceptable quality *
* 0.49 <= DockQ < 0.80 - Medium quality *
* DockQ >= 0.80 - High quality *
* Reference: Sankar Basu and Bjorn Wallner, DockQ:... *
* For comments, please email: bjornw@ifm.liu.se *
***********************************************************
AlphaFold-Multimer的表现:

ipTM
AlphaFold-Multimer 建立了不同链残基之间相互作用的评分系统——Inerface pTM(ipTM),计算类似于pTM,只是分别考虑了不同链(i和j)。

实际使用中,Deepmind采用 pTM 和 ipTM 的加权组合作为模型置信度度量,以便在模型排名中考虑一些链内置信度:


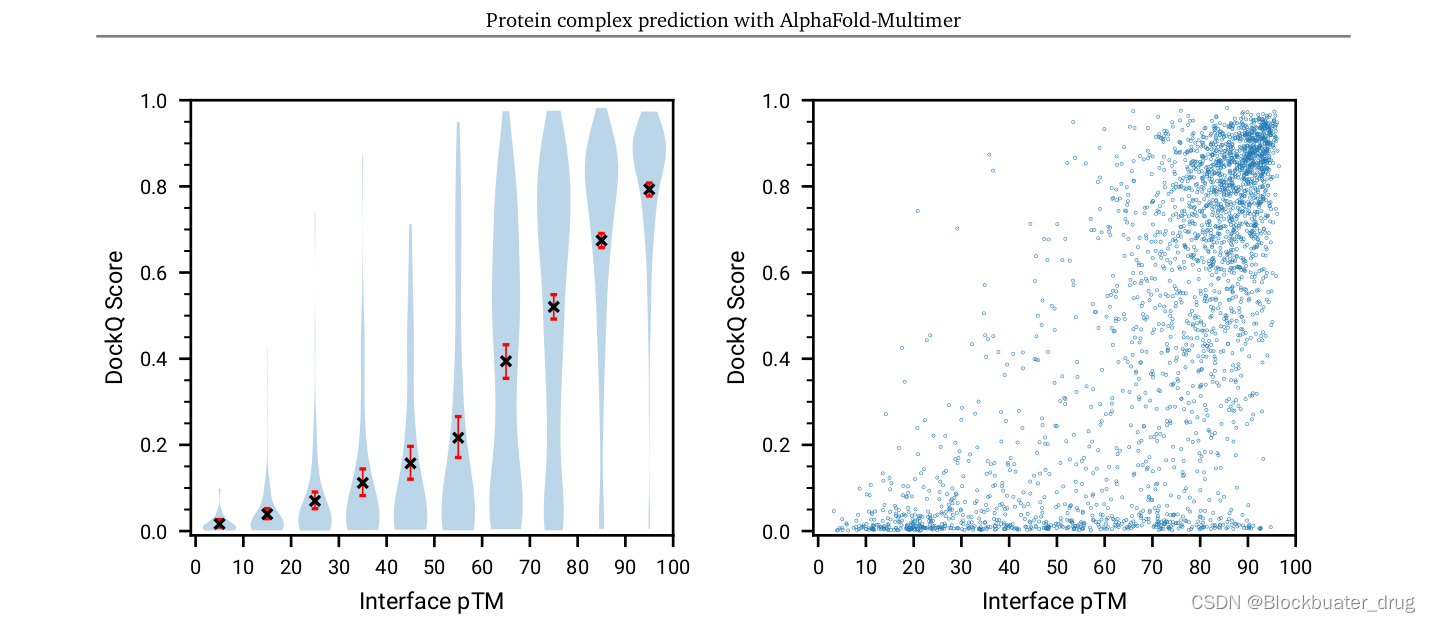 DockQ本身是ipTM的函数,也说明这两个评价multimer的指标具有一定的置信度和准确性。
DockQ本身是ipTM的函数,也说明这两个评价multimer的指标具有一定的置信度和准确性。
总结
以上就是今天的内容,本文汇总了AlphaFold和AlphaFold-multimer等蛋白结构推理预测中,不同蛋白结构预测模型的评价指标。供大家参考。
参考资料
- https://zhuanlan.zhihu.com/p/539157126?utm_id=0
- http://www.360doc.com/showweb/0/0/1113559986.aspx
- https://www.biorxiv.org/content/10.1101/2021.10.04.463034v2.full.pdf
- https://seq2fun.dcmb.med.umich.edu//TM-score/TM-score.pdf
| 欢迎浏览我的CSND博客! Blockbuater_drug …点击进入 |
|---|
这篇关于蛋白结构预测模型评价指标的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!









