本文主要是介绍对77,539个基因组进行的遗传关联分析揭示了罕见疾病的病因,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
今天给同学们分享一篇实验文章“Genetic association analysis of 77,539 genomes reveals rare disease etiologies”,这篇文章发表在Nat Med期刊上,影响因子为82.9。

结果解读:
稀有水库
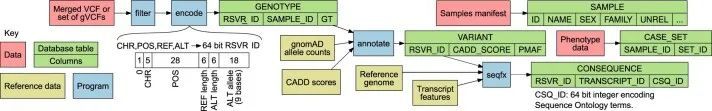
关系型数据库(RDB)提供了一个统一的、集中的结构,用于存储、查询和修改多种底层类型的数据。原则上,RDB可以为基因型、变异、基因、参与者和统计结果的分析提供方便的基础,但它们无法容纳足够规模的表格来存储中等规模队列中的外显子或全基因组基因型。然而,RDB可以容纳仅对应罕见变异的基因型的稀疏表示,这些变异几乎包括对罕见疾病风险产生重大影响的所有变异。作者开发了一个RDB模式,称为Rareservoir,并配套开发了用于罕见疾病分析的构建过程,默认情况下,它存储与所有特定人群MAF可能小于0.1%的变异对应的基因型。这样,在大型研究中,存储的基因型数量可以减少约99%(扩展数据图1)。Rareservoir将变异编码为64位整数('RSVR IDs')(扩展数据图2),可以在实践中表示99.3%的变异而不丢失信息。RSVR ID占据单独的一列,并根据基因组位置按数字递增,使得在简单的数据库结构中可以进行快速的基于位置的查询。为了支持Rareservoir的构建过程,作者开发了一个配套的软件包称为'rsvr'(扩展数据图2和和3)。该软件包包括用于从控制数据库(例如,gnomAD)注释变异的MAF信息的工具,致病性评分(例如,组合注释依赖耗尽(CADD)评分)和与一组转录本相关的预测序列本体(SO)后果。作者使用64位整数('CSQ ID')来记录相互作用的变异/转录本对的后果,其中每个位编码一种可能的后果,按严重性排序。以这种方式编码后果是高效的,并且可以进行简洁的查询,根据影响的严重程度进行阈值或排序。Rareservoir还包含一个表格,其中包含每个样本的遗传衍生数据(包括血统、性别和最大无关参与者集的成员资格),以及一个存储每个参与者所分配的罕见疾病类别的“病例集”表格。

BeviMed推断出241个已知和19个未知的遗传关联
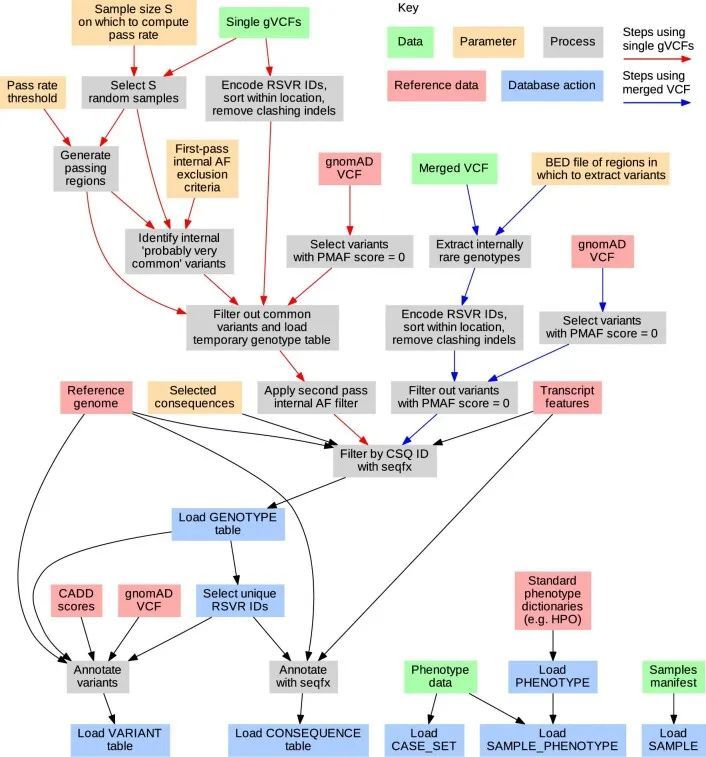
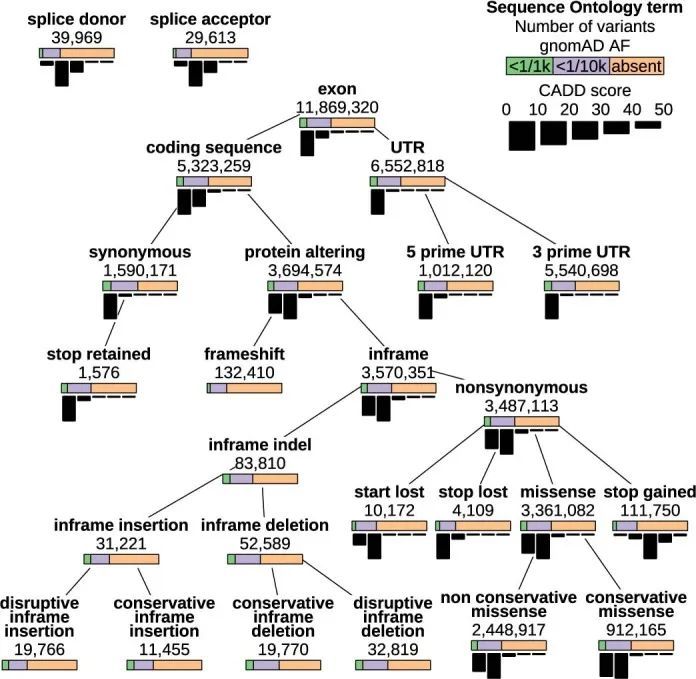
作者建立了一个大小为5.5 GB的Rareservoir,其中包含了Ensembl v.104中影响蛋白编码基因的经典转录本的1190万个罕见外显子和剪接单核苷酸变异(SNV)以及短插入或缺失(indels)。这些变异来自于一个合并的变异呼叫格式文件(VCF),其中包含了77,539名参与者(包括29,741名先证者)在100KGP(数据发布v.13)的罕见疾病主要计划中的基因型呼叫。在加入100KGP时,专家临床医生根据每个受影响参与者的临床特征将其分配到220个“特定疾病”中的一个或多个。这些特定疾病按层次排列为88个“疾病子组”,每个子组属于20个“疾病组”之一。虽然许多特定疾病的资格标准与相同或密切相关的罕见疾病相一致,但对于其他疾病(如“智力障碍”)的标准更广泛,涵盖了多种遗传病因。作者生成了269个分析案例集,对应于所有不同的具体疾病和疾病亚组,大小从5,809到一个先证者不等,并将它们存储在Rareservoir中(图1a和扩展数据图5和6)。作者包括了这两个表型层次,以考虑在共享相同遗传病因的病例中表现或诊断的异质性,旨在提高识别统计遗传关联的能力。



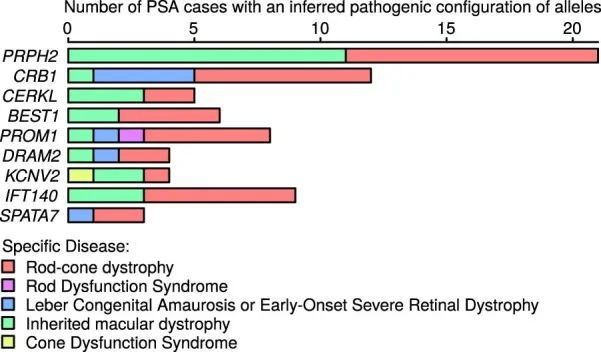
使用贝叶斯统计方法BeviMed,作者得到了19,663个蛋白编码基因与269个罕见疾病类别之间的后验概率(PPA)。BeviMed计算了在没有关联的基线模型和竞争关联模型之间的后验概率,每个模型都假设了特定的遗传方式(MOI;显性或隐性)和病因变异的后果类别(在本研究中为高影响、中等影响或5'非翻译区(UTR))。通过对所有关联模型的后验概率求和,得到了PPA。具有最大后验概率的关联模型(模态模型)确定了推断的MOI和病因变异的类别。在关联模型的条件下,BeviMed对每个包含的罕见变异的致病性进行建模。在该模型中,具有至少一个致病等位基因(在显性MOI下)或至少与倍性相同数量的致病等位基因(在隐性MOI下)的参与者具有致病等位基因的配置,这决定了他们的病例状态风险。对于每个罕见疾病类别,作者根据100KGP提供的家系信息选择了一组无关的病例,并将它们与不属于病例组的来自不同家系和由100KGP提供的一组最大的无关参与者进行比较。为了考虑病例组之间的相关性,作者只记录了在给定疾病组中具有最高PPA的每个基因的关联。使用PPA > 0.95的显著性阈值,作者确定了260个显著的关联,其中241个由PanelApp基因面板数据库 14 记录,这是一个由专家策划和注释的资源,包含具有罕见疾病因果性高、中或低先前支持证据的基因列表(图1b)。在作者确定的241个已知关联中,有43个(17.8%)与疾病亚组有关。例如,在与“后部段异常”疾病亚组相关的九个已知基因中,由具有条件致病性后验概率 > 0.8的变异解释的病例集包括涵盖多种具体疾病的参与者(扩展数据图7)。这表明,属于同一疾病亚组的不同特定疾病的参与者有时会在同一基因中共享缺陷,这证实了将疾病亚组而不仅仅是特定疾病作为案例集来处理可以提高统计能力。
ERG基因的变异导致原发性淋巴水肿
BeviMed发现了ERG基因中高影响变异体与特定疾病“原发性淋巴水肿”之间的显性遗传关联,这是一组由淋巴管异常发育或淋巴功能失调引起的遗传疾病。其中三种变异体负责高PPA,位置范围从编码182到463的Ensembl转录本ENST00000288319.12上。其中一个患者的两个未受影响的父母都没有这种变异等位基因,一个通过100KGP测序,另一个通过Sanger测序,这表明这种截短的杂合变异体是新出现的。第四个家庭的一个参与者因为与无关疾病而被纳入100KGP,但他也携带了ERG基因中预测的功能丧失变异体。经过手动图表审核,发现该参与者具有与该无关疾病相关的特征,但也有与原发性淋巴水肿一致的额外特征,这在发现队列中提供了内部复制(图2a)。
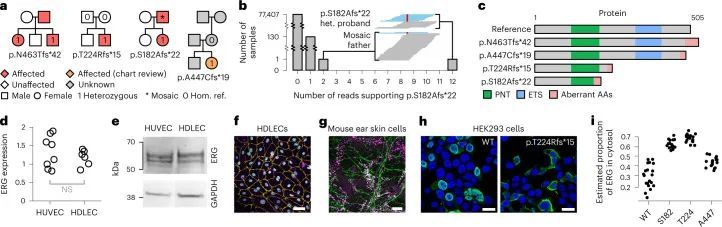
患有编码p.S182Afs*22变异的患者的受影响父亲被称为纯合子参考等位基因,最初暗示该变异与该家系中的疾病不共分离。然而,对父亲的GS读取对齐进行审查后发现,在该位置上有48个读取与该替代等位基因相符。具体而言,这些读取包含在‘AGCTGGGGGTGAG’模体的中央多G序列中删除了一个G。为了评估这是否可能是错误测序的结果,作者统计了100KGP中77,539个基因组中具有这种读取的数量,并发现患者和父亲是仅有的两个具有多个这种读取的人。这表明父亲的这些读取不太可能是错误的,而是他是嵌合体(图2b),与他的淋巴水肿比他的女儿晚出现二十多年的临床观察一致,表明疾病较轻。通过100KGP收集的另外130个样本中有一个包含该删除的读取。这个数字与其他80个包含相同13个碱基对(bp)基序的外显子位点的观察结果一致(平均99.67个样本,范围为4-149个样本),这表明这130个样本不是镶嵌的,而是包含了个别测序错误。此外,给出这些样本的参与者中没有一个被指定为“原发性淋巴水肿”特定疾病。
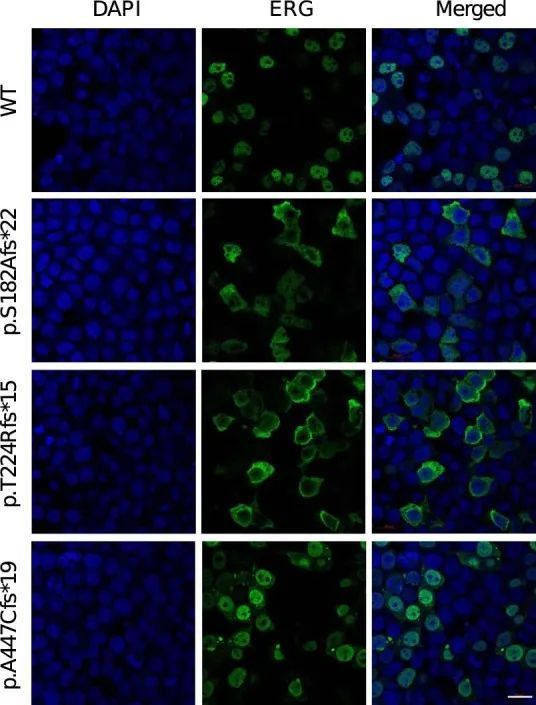
ERG编码了血管内皮细胞基因表达的关键转录调节因子,对正常血管发育至关重要。然而,关于ERG对淋巴发育的贡献以及如何通过影响ERG蛋白的不同部分而导致功能丧失的ERG变异体引起原发性淋巴水肿的机制知之甚少(图2c)。通过实时定量聚合酶链反应(PCR)检测纯化RNA和免疫印迹蛋白提取物,发现ERG在原发性人皮淋巴内皮细胞(HDLECs)和人脐静脉内皮细胞(HUVEC)中的总细胞表达相同(分别见图2d、e)。此外,培养的HDLECs的免疫荧光显微镜观察显示,ERG表达与淋巴内皮细胞核标记物PROX1共定位(图2f),这一发现在小鼠出生后3周的耳部皮肤整体染色图中得到了证实(图2g)。p.S182Afs*22和p.T224Rfs*15变异体的位置提示可能存在无义介导的降解和单等位基因不足作为潜在的疾病机制。然而,另外两种变体位于ERG的最后一个外显子中,因此可能逃避无义介导的降解。作者对这两种变体进行了更详细的研究,以探索潜在的疾病机制。在不表达内源性ERG的HEK293细胞中,过表达野生型ERG cDNA重现了在HDLEC和小鼠耳皮肤模型中观察到的核表达模式。然而,ERG突变型cDNA的过表达导致ERG在细胞核外的胞质中定位错误(图2h,i和扩展数据图8),阻止其与DNA结合并发挥作为转录因子的功能。综上所述,这些数据确认了淋巴内皮细胞核中ERG表达水平较高,与淋巴管生成过程中的转录调控功能一致。它们还表明,在原发性淋巴水肿病例中,缺陷的淋巴管生成可能是由于核内ERG可用性减少,要么是由于无义介导的降解导致的半失能,要么是由于定位错误所致。
PMEPA1基因变异导致洛伊斯-迪茨综合征
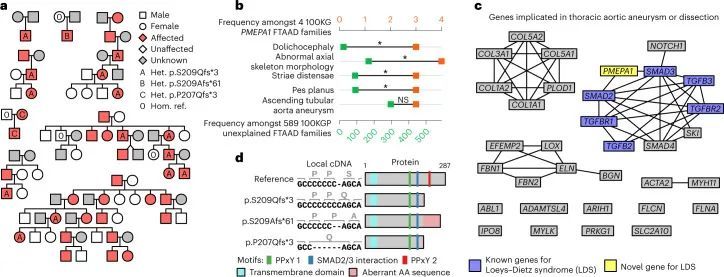
BeviMed发现了PMEPA1基因中高影响变异体与特定疾病“家族性胸主动脉瘤病”(FTAAD)之间的显性遗传关联。具有最高条件致病性概率的变异体是在规范Ensembl转录本ENST00000341744.8的最后一个外显子中的七个胞嘧啶序列中插入一个胞嘧啶。这个变异体被预测会引起p.S209Qfs*3的移码突变,在100KGP发现队列的三个欧洲血统FTAAD家系中观察到。作者在另外三个病例集合中复制了这个关联。首先,在一个独立的日本患者群体中,作者独立地发现了相同的变异体,该变异体在三个日本血统家系的八名患者中被发现。其次,在100KGP Pilot Programme的一个独立的2,793名参与者集合中,作者发现了与前一个变异体相同的多胞嘧啶序列中的单个胞嘧啶缺失,编码为p.S209Afs*61,该患者为FTAAD病例。最后,作者在比利时发现了一个家庭,受影响的成员携带了同一段多胞嘧啶中的5个碱基缺失,导致其他两个变异体的上游两个残基发生了移码突变(p.P207Qfs*3)。
所有家系都表现出主导遗传的主动脉瘤病,但穿透率不完全,并伴有胸骨畸形、脊柱侧弯和蜘蛛指等骨骼特征,穿透率完全,与基因分型参与者中的相应变异共分离(图3a)。为了评估受FTAAD影响的PMEPA1家族是否形成了表型上有别的亚组,作者分析了100KGP两个计划中分配给593个FTAAD家族的人类表型本体(HPO)术语。使用基于Resnik等人的语义相似度测量的基于排列的方法,作者发现四个100KGP的PMEPA1家族之间的相似度显著高于随机选择的其他FTAAD家族(P = 5.7 × 10)。为了更详细地描述PMEPA1表型,作者比较了至少在四个家族中存在的最小一组术语中每个HPO术语的患病率与其他FTAAD家族的患病率。作者发现与肌肉骨骼系统相关的四个HPO术语显著富集(图3b), 回应了Loeys-Dietz综合征的表型特征。
GPR156的变异导致隐性先天性听力损失
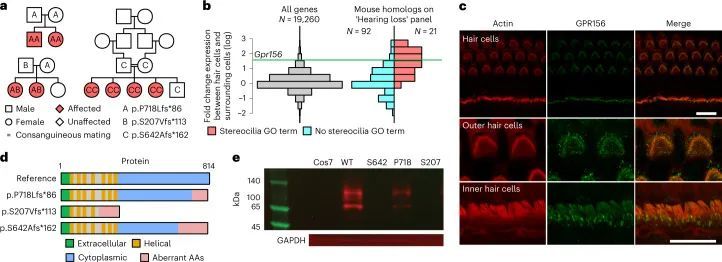
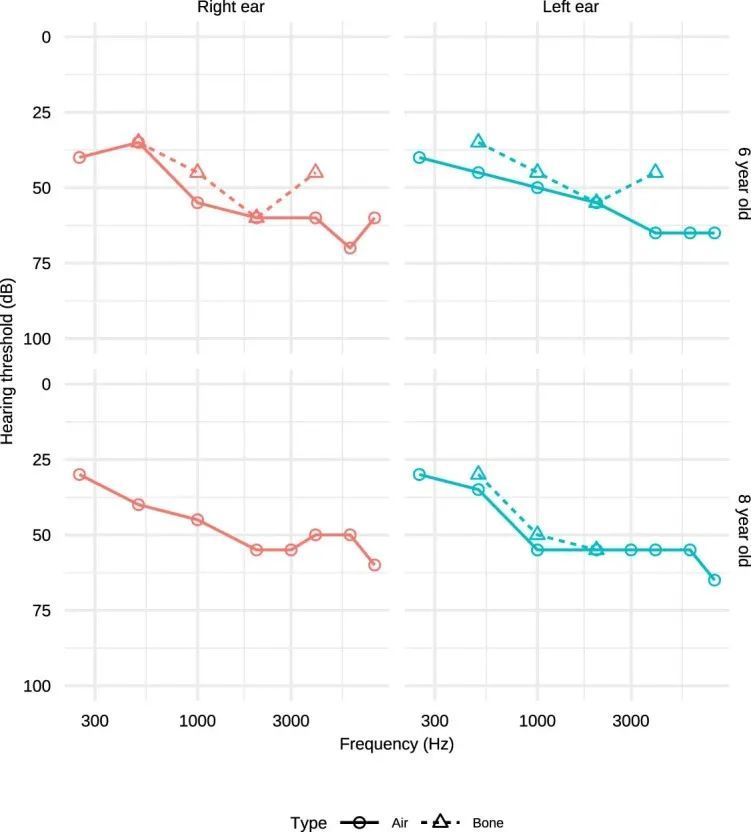
BeviMed发现了GPR156基因中高影响变异与特定疾病“先天性听力障碍”之间的隐性遗传关联。GPR156中的两个高影响变异负责了关联的强有力证据:一个1个碱基插入预测p.S207Vfs*113,以及一个1个碱基插入预测p.P718Lfs*86,相对于规范的Ensembl转录本ENST00000464295.6。一个家庭包含两个受影响的兄弟姐妹,他们都是由杂合父母遗传的p.S207Vfs*113变异的纯合子。在第二个家庭中,也有两个受影响的兄弟姐妹,这种情况下是由母亲遗传的相同的p.S207Vfs*113变异和父亲遗传的不同的p.P718Lfs*86变异的复合杂合子。使用GeneMatcher,作者在沙特阿拉伯发现了第三个家系,其中GPR156中存在双等位截短变异。这个近亲家系包含四个患有听力障碍的兄弟姐妹,他们都是一个预测p.S642Afs*162的变异的纯合子(图4a)。这三个家庭中的八个受影响个体都患有先天性非综合征性双侧感音神经性听力损失(见扩展数据图)。99个用于说明的听力图谱。
总结
GS在医疗系统内的标准化,以及对遗传和表型数据处理和统计分析的强大框架,有望推动对罕见疾病尚未解明的病因的进一步研究。作者开发了一种轻量级且易于部署的RDB(Rareservoir),用于使用BeviMed等方法对罕见疾病进行遗传分析。在一个统一的分析中,作者发现了260个关联,其中241个在之前的几十年的遗传学研究中已经发表过。作者的结果给出了7.3%的误发现率的上限。相比之下,最近对100KGP中的57,000个样本进行的分析报告了249个已知的和579个以前未知的关联,误发现率的上限为70%,这表明作者的分析方法在给定敏感性下具有更高的特异性。这些关联涵盖了86个疾病类别,涉及广泛的器官系统。
这篇关于对77,539个基因组进行的遗传关联分析揭示了罕见疾病的病因的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!






