本文主要是介绍RNA-seq分析:Step9(共表达分析),希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
目录
前记
一、R包的安装
二、数据的载入和预处理
1、数据导入
2、样本的聚类
3、软阈值的计算和选择
三、生成基因模块
四、基因模块与表型之间联系
五、导出共表达网络文件
后记
前记
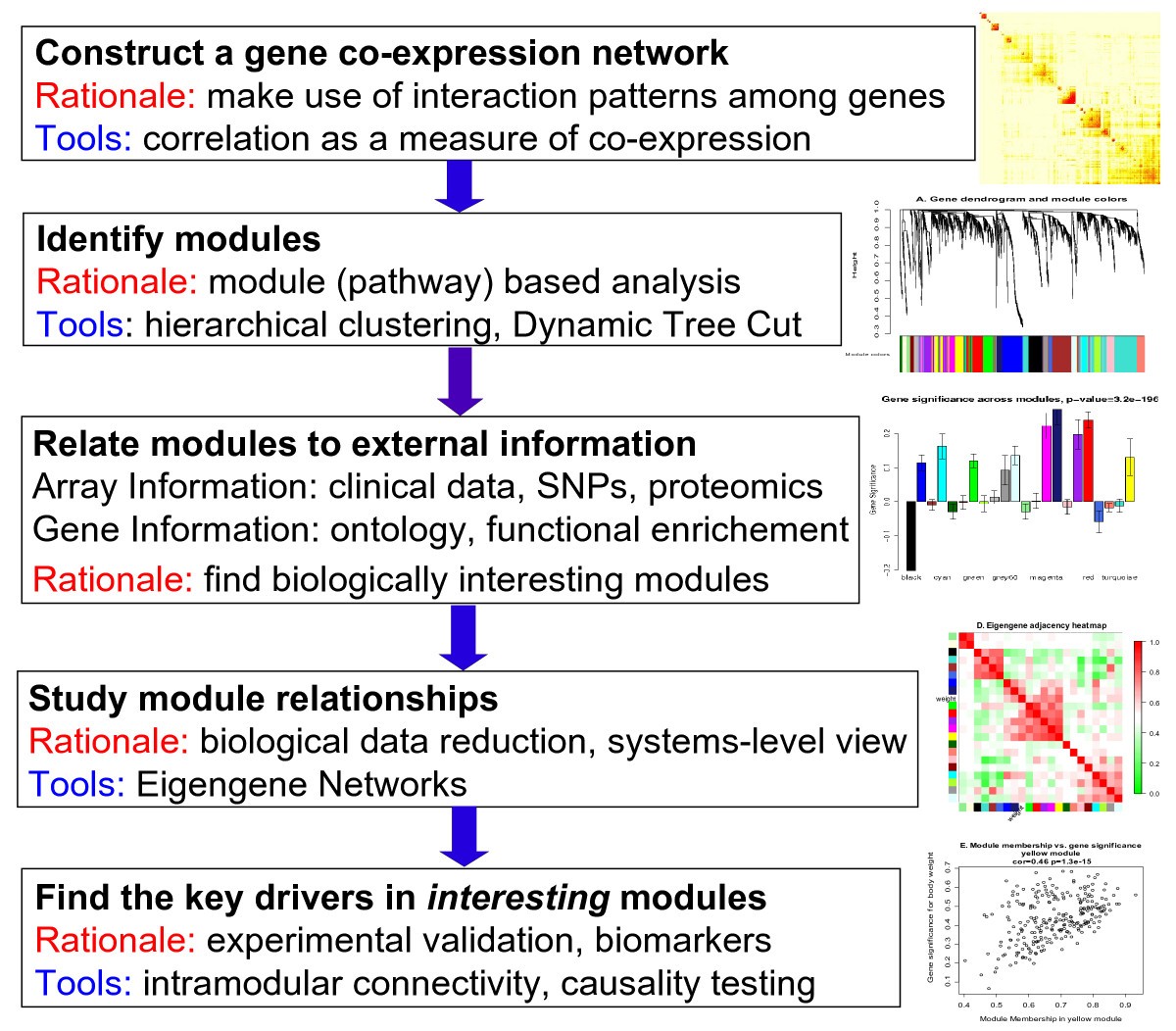
WGCNA(Weighted Correlation Network Analysis)是一种系统生物学中常用的数据分析方法,主要用于分析高通量基因表达数据。该方法通过构建基因共表达网络,将相似的基因组织到同一模块中,并用模块间的关联性进行分析,从而识别与生物学过程相关的模块和关键基因。
WGCNA分析流程主要包括:数据预处理、构建共表达网络、模块检测、模块注释和功能分析等步骤。其中,构建共表达网络是WGCNA的核心步骤,其基本思想是通过计算基因间的相关系数,建立基因共表达网络,然后利用模块检测算法将相关基因聚类成模块,进而探究不同模块与生物学过程的相关性。最终,可以对模块进行注释和功能分析,揭示其在生物学过程中的作用。
一、R包的安装
WGCNA分析需要用到WGCNA包。WGCNA R包是基于R语言编写的,用于实现WGCNA分析的工具包。该包提供了一系列函数,可用于数据预处理、构建基因共表达网络、模块检测、模块注释和功能分析等步骤。同时,还提供了数据导入和导出、可视化等功能。
WGCNA R包中的主要函数包括:
- importDataset:用于导入基因表达数据;
- goodSamplesGenes:用于筛选高质量的样本和基因;
- pickSoftThreshold:用于选择共表达网络分析中的软阈值;
- blockwiseModules:用于构建基因共表达网络,并将相关基因聚类成模块;
- moduleColors:用于将模块标记颜色并可视化;
- moduleTraitCor:用于计算模块和外部性状的相关性;
- pathwayAnalysis:用于对模块进行通路分析。
WGCNA R包是一个功能强大的工具,可以快速、高效地进行WGCNA分析。对于想要进行基因表达数据分析的生物学家和生物信息学家来说,是一个非常有价值的工具。
WGCNA: an R package for weighted correlation network analysis | BMC Bioinformatics | Full Text (biomedcentral.com)![]() https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-9-559#Fig1安装的代码如下:
https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-9-559#Fig1安装的代码如下:
#载入WGCNA和flashClust
BiocManager::install("impute")
BiocManager::install("preprocessCore")
install.packages("WGCNA")
install.packages("flashClust")
library(WGCNA)
library(flashClust)二、数据的载入和预处理
1、数据导入
导入的数据包括差异基因的表达量信息和性状信息,代码如下:
#读入表达量数据
fpkm <- read.table("GSE80565_RNAseq_ABAtimeSeries_rpkm_log2FC_FDR_filterCPM2n2.txt", header = TRUE)
rownames(fpkm) <- fpkm$gene_id #设置第一列为列名
fpkm <- fpkm[, - 1] #删除第一列
fpkm <- fpkm[, 1:28] #提取表达量数据
fpkm <- log2(fpkm + 1) #取对数#读入差异表达分析结果
deseq_results <- read.csv("DESeq2_results_significant.csv", row.names = "X", header = TRUE)
SubGeneNames <- as.character(rownames(deseq_results)) #提取差异表达基因名称
datExpr <- fpkm[SubGeneNames, ] #提取差异表达基因表达量数据
datExpr <- na.omit(datExpr) #去除NA值
datExpr <- as.data.frame(t(datExpr)) #颠换行和列#检查是否存在异常值
gsg <- goodSamplesGenes(datExpr, verbose = 3)
gsg$allOK#读入表型数据
datTraits <- read.table("traits_wgcna.txt")
#datTraits <- datTraits[1:14, ]
table(rownames(datTraits) == rownames(datExpr))2、样本的聚类
#根据表达量对样本进行聚类
A <- adjacency(t(datExpr), type = "signed")
k <- as.numeric(apply(A, 2, sum)) - 1
Z.k <- scale(k)
thresholdZ.k <- - 2.5
outlierColor <- ifelse(Z.k < thresholdZ.k, "red", "black")
sampleTree <- flashClust(as.dist(1 - A), method = "average")#样本聚类树(red indicates high values)
traitColors <- data.frame(numbers2colors(datTraits, signed = FALSE))
dimnames(traitColors)[[2]] <- paste(names(datTraits))
datColors <- data.frame(outlier = outlierColor, traitColors)
plotDendroAndColors(sampleTree, groupLabels = names(datColors), colors = datColors, main = "Sample Dendrogram and Trait Heatmap")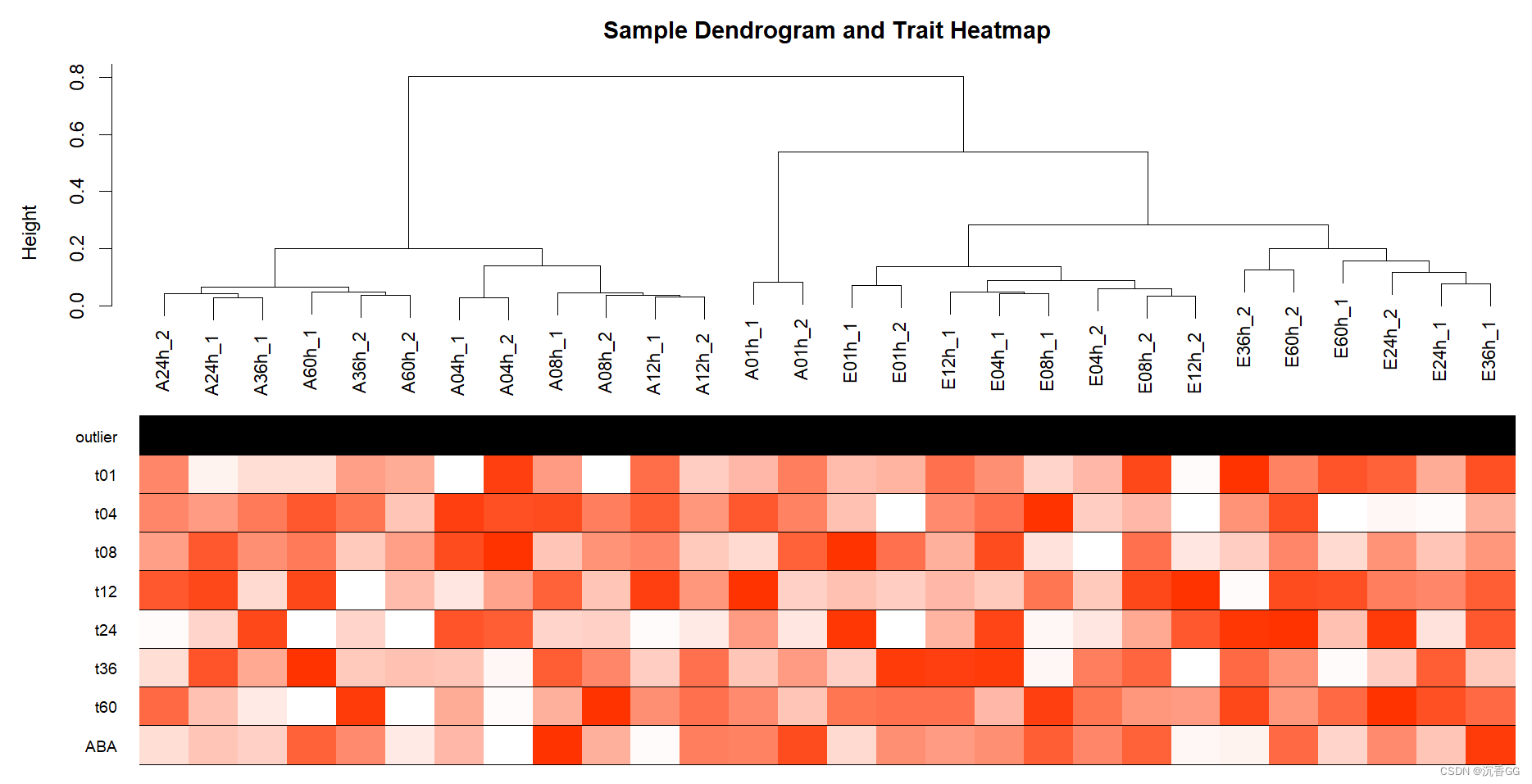
3、软阈值的计算和选择
在WGCNA分析中,软阈值是选择共表达基因模块时最为关键的参数之一。软阈值是将基因表达数据矩阵中每个基因与其它基因的相关性系数的绝对值进行幂指数化,以使得相关性系数呈现幂律分布。这个幂指数化参数就是软阈值。软阈值越大,相似性系数低的基因对会被更加压抑,而相似性系数高的基因对则会被更加放大,这样可以使得基因之间的相关性更加明显。
WGCNA通常会构建一个基因相关系数矩阵,该矩阵可以通过计算基因间的Pearson相关系数、Spearman相关系数或其他相关系数来获得。然后思考如何将这些基因聚集成基因模块,通常情况下,会使用类似于层次聚类的方法将相似的基因聚集在一起,并根据特定的指标为每个模块分配一个标记,这个过程称为分组。
选择适当的软阈值对于分组结果的准确性和稳定性至关重要。通常可以使用WGCNA软件中自带的函数“pickSoftThreshold”来自动计算软阈值,该函数会根据基因表达数据的特性自动选择最佳的软阈值。当然,也可以手动调整软阈值,观察不同软阈值对模块划分影响的变化,找到一个合适的软阈值。
#选择恰当的幂次
powers <- c(c(1:10), seq(from = 12, to = 60, by = 2))
sft <- pickSoftThreshold(datExpr, dataIsExpr = TRUE, powerVector = powers, corFnc = cor, corOptions = list(use = 'p'), networkType = "signed")#Plot softPower results
par(mfrow = c(1, 2))
cex1 = 0.9#Scale-free topology fit index as a function of the soft-thresholding power
plot(sft$fitIndices[, 1], -sign(sft$fitIndices[, 3])*sft$fitIndices[, 2], xlab = "Soft Threshold (power)", ylab = "Scale Free Topology Model Fit, signed R^2", type = "n", main = paste("Scale independence"))
text(sft$fitIndices[, 1], -sign(sft$fitIndices[, 3])*sft$fitIndices[, 2], labels = powers, cex = cex1, col = "red")
abline(h = 0.80,col = "red")#Mean connectivity as a function of the soft-thresholding power
plot(sft$fitIndices[, 1], sft$fitIndices[, 5], xlab = "Soft Threshold (power)", ylab = "Mean Connectivity", type = "n", main = paste("Mean connectivity"))
text(sft$fitIndices[, 1], sft$fitIndices[, 5], labels = powers, cex = cex1, col = "red")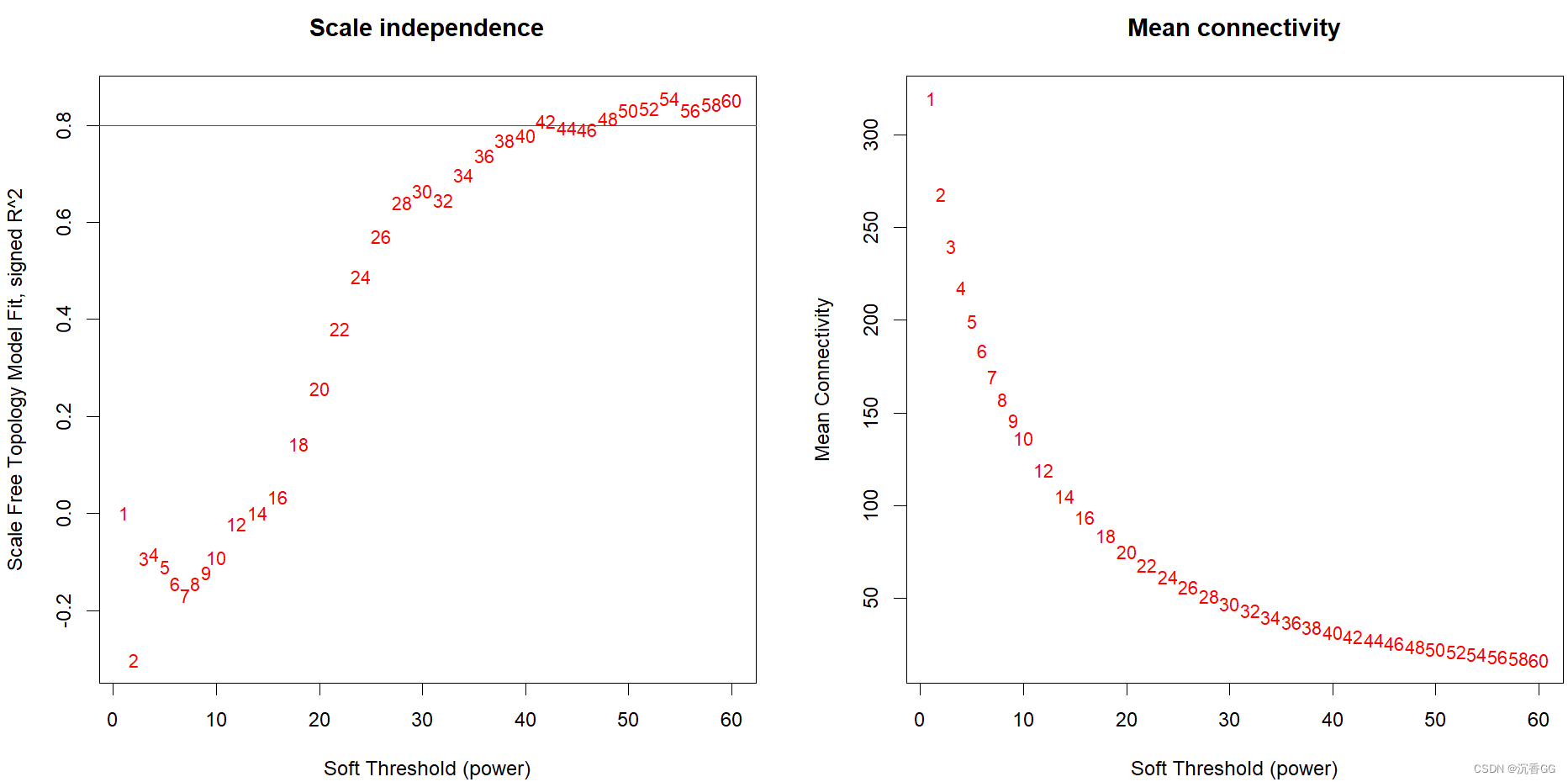
补充1:
作为软阈值功率函数的无标度拓扑拟合指数指的是一种网络拓扑的度量方式,用于衡量一个网络的无标度性质,即网络中节点的度数分布服从幂律分布。该指数采用软阈值函数对幂律分布进行拟合,从而得到能够描述网络无标度性质的指数。具体来说,软阈值函数在拟合幂律分布时可以减少高度不可靠的极端值的影响,提高模型拟合的准确性。因此,使用软阈值功率函数作为拟合函数的无标度拓扑拟合指数可以更准确地描述网络的无标度性质。
补充2:
无标度拓扑拟合指数(又称无标度网络指数或无标度拓扑标准)是用于评估网络是否遵循无标度拓扑的一种度量方法,无标度拓扑的特点是有少数高度连接的节点(枢纽)和许多稀疏连接的节点。该指数通过将网络的度分布拟合为幂律分布来计算。
软阈值功率是一个参数,用于根据基因表达数据集构建加权邻接矩阵。该参数用于确定基因间相关性的强度,并过滤掉可能与生物无关的较弱相关性。
随着软阈值处理能力的变化,生成的网络可能会表现出不同程度的无标度性。因此,作为软阈值功率函数的无标度拓扑拟合指数,可以评估在不同强度阈值下,所得到的网络与无标度拓扑的吻合程度。这一点非常重要,因为不同的生物系统可能有不同程度的无标度,了解特定数据集的最佳阈值可以提高基因共表达网络分析等下游分析的准确性。
三、生成基因模块
WGCNA(Weighted Gene Co-Expression Network Analysis)是一种用于分析基因共表达网络的方法,它能够将大量的基因数据转化为共表达模块,从而为后续分析提供便捷。以下是WGCNA分析生成基因模块的简述:
-
数据预处理:首先需要进行数据预处理,包括基因表达数据质控、归一化、去除批次效应等步骤。
-
构建共表达网络:通过计算基因之间的相关性,构建基因共表达网络。可以使用Pearson相关系数、Spearman相关系数等方法来计算基因之间的相关性。
-
模块划分:将基因按照其相似性聚类成若干个模块。可以使用层次聚类、动态树切割等算法来实现。
-
模块注释:对每个模块进行注释,可以使用基因富集分析等方法,了解每个模块所代表的生物学功能和通路。
-
模块与表型关联:将每个模块的表达量与不同表型数据进行相关分析,找到与表型显著相关的模块。
-
模块提取:最终选取与表型显著相关的模块,得到这些模块中包含的基因列表,这些基因就可以作为后续分析的重要基础。
enableWGCNAThreads() softPower <- 56 adjacency <- adjacency(datExpr, power = softPower, type = "signed")#生成TOM矩阵,计算相异性 TOM <- TOMsimilarity(adjacency, TOMType = "signed") dissTOM <- 1 - TOM#基因聚类树 geneTree <- flashClust(as.dist(dissTOM), method = "average") par(mfrow = c(1, 1)) plot(geneTree, xlab = "", sub = "", main = "Gene Clustering on TOM-based dissimilarity", labels = FALSE, hang = 0.04)minModuleSize <- 15 #设置模块至少包含的基因数dynamicMods <- cutreeDynamic(dendro = geneTree, distM = dissTOM, deepSplit = 2, pamRespectsDendro = FALSE, minClusterSize = minModuleSize) dynamicColors <- labels2colors(dynamicMods) MEList <- moduleEigengenes(datExpr, colors = dynamicColors, softPower = softPower) MEs <- MEList$eigengenes MEDiss <- 1 - cor(MEs) METree <- flashClust(as.dist(MEDiss), method = "average")#模块聚类树 plot(METree, main = "Clustering of module eigengenes", xlab = "", sub = "") #设置合并模块阈值 MEDissThres <- 0.00 #not to merge merge <- mergeCloseModules(datExpr, dynamicColors, cutHeight = MEDissThres, verbose = 3) mergedColors <- merge$colors mergedMEs <- merge$newMEs#基因聚类树以及模块划分 plotDendroAndColors(geneTree, cbind(dynamicColors, mergedColors), c("Dynamic tree cut", "Merged dynamic"), dendroLabels = FALSE, hang = 0.03, addGuide = TRUE, guideHang = 0.05) moduleColors <- mergedColors colorOrder <- c("grey", standardColors(50)) moduleLabels <- match(moduleColors, colorOrder) - 1 MEs <- mergedMEs#模块间基因连通性值计算并排序 allDegrees <- intramodularConnectivity(adjacency, dynamicColors) allDegrees <- allDegrees[order(allDegrees$kTotal), ] allDegrees <- data.frame(GeneID = rownames(allDegrees), allDegrees) write.table(allDegrees, "degrees.txt", sep = "\t", quote = FALSE, row.names = FALSE)#提取模块基因,输出至文件 module_colors = setdiff(unique(dynamicColors), "grey") for(color in module_colors){module_genes = SubGeneNames[which(dynamicColors == color)]write.table(module_genes, paste("module_", color, ".txt", sep=""), sep="\t", row.names=FALSE, col.names=FALSE, quote=FALSE) }#TOM图 #set the diagonal of the dissimilarity to NA diag(dissTOM) = NA; #Visualize the Tom plot. Raise the dissimilarity matrix to a power to bring out the module structure sizeGrWindow(7,7) TOMplot(dissTOM^4, geneTree, as.character(dynamicColors))
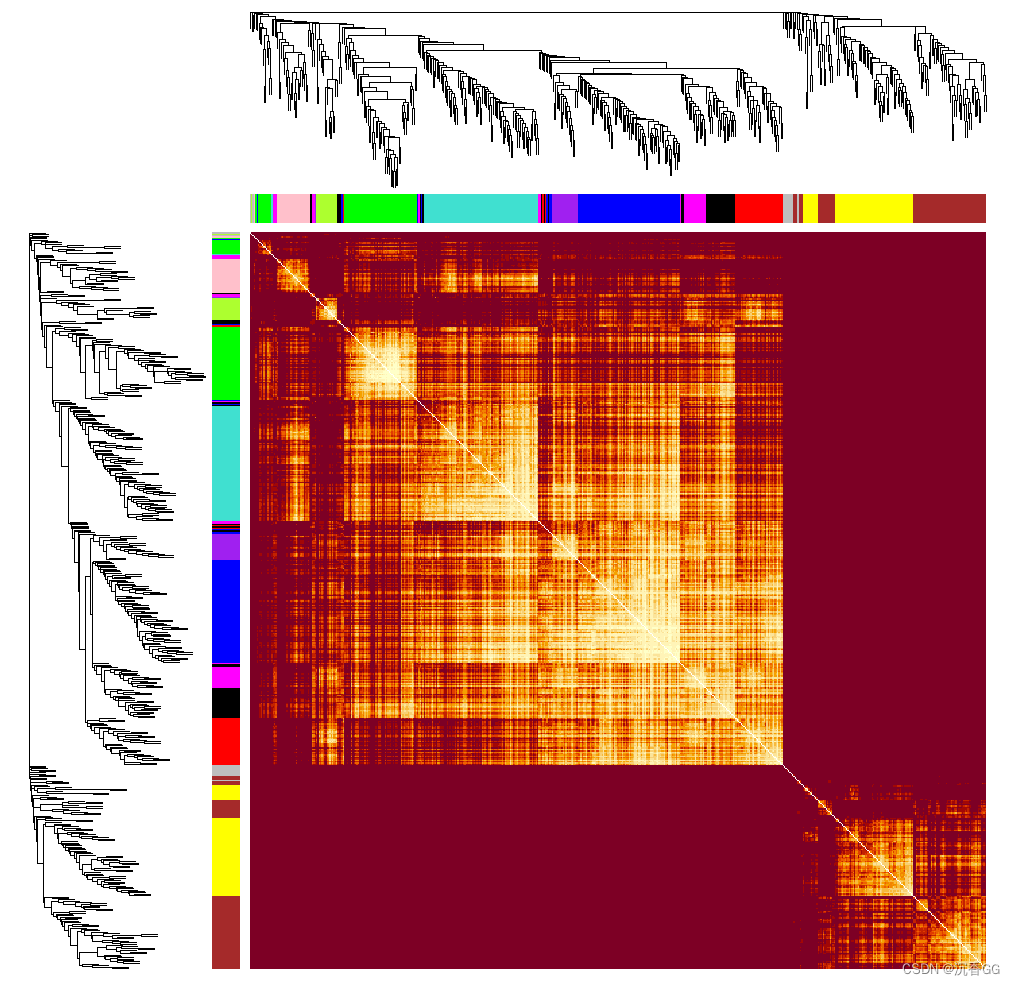
WGCNA(Weighted Gene Co-Expression Network Analysis)是一种常用的基因共表达网络分析方法,TOM(Topological Overlap Matrix)图是WGCNA分析中最常见和重要的图表之一。下面是解读WGCNA分析中TOM图的一般步骤:
1. TOM图是什么?TOM图是基于WGCNA分析构建的基因共表达网络图,它显示了基因和基因之间的“拓扑重叠”程度,即基因之间的相似性或连接强度。
2. TOM图如何解读?TOM图通常包含两个关键参数:基因列表和TOM值。基因列表是标识网络中每个节点(基因)的唯一标识符,TOM值表示“拓扑重叠”程度,数值越高表示基因之间的相似性或连接越强。
3. 颜色模块:为了更好地表示基因之间的相似性和连接强度,通常将TOM值相近的基因进行聚类,形成不同的颜色模块。这些颜色模块可以反映出共表达基因的生物学功能和通路。
4. 颜色条:颜色条是显示每个基因在不同颜色模块中的位置的横向条形图,颜色条越长表示该基因被聚类在多个模块中的概率越高。
5. 热图:为了更方便地查看基因之间的连接程度,通常会使用热图展示基因与基因之间的TOM值,颜色越深表示TOM值越大,即连接越强。
综上,通过解读TOM图可以了解到基因共表达网络中基因之间的相似性和连接强度,以及不同颜色模块所代表的生物学功能和通路。
四、基因模块与表型之间联系
WGCNA(Weighted Gene Co-expression Network Analysis)是一种常用于分析基因表达数据的方法,它可以通过构建基因共表达网络来鉴定与疾病相关的基因模块,并探索这些基因模块与表型的关联性。在WGCNA分析中,基因模块通常被定义为具有高度相关性的基因组成的子集,这些基因模块可以反映生物学过程或预测某些重要的表型。
对于基因模块与表型之间的联系,可以通过计算模块特异性的基因表达量和表型数据之间的相关性来进行评估。这种关联性分析可以帮助我们发现哪些基因模块与表型有关,从而更好地理解基因组学与表型之间的联系。同时,WGCNA还可以通过对基因模块进行富集分析,来鉴定这些模块与特定生物学过程或疾病有关的基因,并提供更深入的生物学信息。
总之,WGCNA分析可以帮助我们鉴定基因模块,并探索这些模块与表型之间的联系,从而提高我们对基因组学与表型之间关系的了解。
#Define number of gengs and samples
nGenes <- ncol(datExpr)
nSamples <- nrow(datExpr)#Recalculate MEs with color labels
MEs0 <- moduleEigengenes(datExpr, moduleColors)$eigengenes
MEs <- orderMEs(MEs0)
moduleTraitCor <- cor(MEs, datTraits, use = 'p')
moduleTraitPvalue <- corPvalueStudent(moduleTraitCor, nSamples)#Print correlation heatmap between modules and traits
textMatrix <- paste(signif(moduleTraitCor, 2), "\n(", signif(moduleTraitPvalue, 1), ")", sep = "")
dim(textMatrix) <- dim(moduleTraitCor)#Display the correlation value with a heatmap plot
sizeGrWindow(12, 9)
par(mar = c(6, 8.5, 3, 3))
labeledHeatmap(Matrix = moduleTraitCor,xLabels = names(datTraits),yLabels = names(MEs),ySymbols = names(MEs),colorLabels = FALSE,colors = blueWhiteRed(50),textMatrix = textMatrix,setStdMargins = FALSE,cex.text = 0.5,zlim = c(-1, 1),main = paste("Module-trait relationships"))
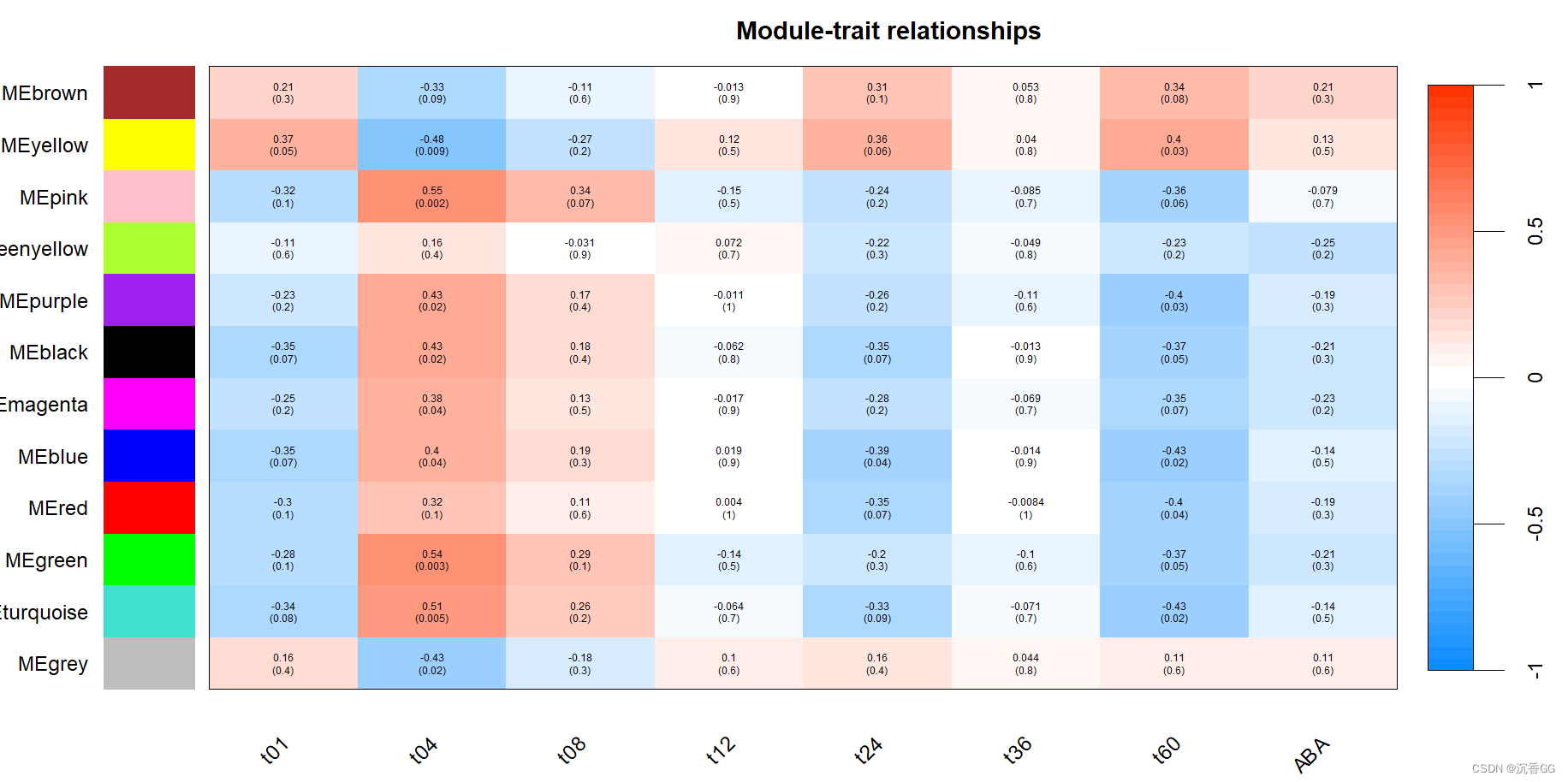
五、导出共表达网络文件
共表达网络文件是指包含基因表达数据的格式化文件,以及通过这些数据构建的基因共表达网络。在WGCNA分析中,共表达网络文件通常包含两部分内容:
-
基因表达数据:基因表达数据是指在不同生理条件下的基因表达水平,通常以矩阵的形式呈现。基因表达数据可以通过RNA测序或基因芯片技术获得。其中,行表示基因,列表示不同的样本,每个元素表示该基因在该样本中的表达水平。
-
权重矩阵:权重矩阵是通过基因表达数据计算得到的共表达网络矩阵。该矩阵中的元素表示两个基因之间的相似性,通常以Pearson相关系数或Spearman相关系数的值表示。共表达网络的构建需要对基因表达数据进行预处理(例如,去除批次效应、标准化等),并调整参数(例如,软阈值)以得到较好的共表达模块。
共表达网络文件可以使用多种方式进行可视化和分析,例如通过Cytoscape软件进行网络可视化,或使用R语言中的WGCNA包进行模块鉴定和生物学意义富集分析等。这些分析可以帮助我们理解基因共表达网络中的生物学意义,从而为生物学研究提供更深入的信息和启示。
modules <- setdiff(unique(dynamicColors), "grey")
genes <- names(datExpr)
inModule <- is.finite(match(moduleColors, modules))
modProbes <- genes[inModule]
modGenes <- genes[inModule]
modTOM <- TOM[inModule, inModule]
dimnames(modTOM) <- list(modGenes, modGenes)cyt = exportNetworkToCytoscape(modTOM,edgeFile = paste("CytoscapeInput-edges-", paste(modules, collapse="-"), ".txt", sep=""),nodeFile = paste("CytoscapeInput-nodes-", paste(modules, collapse="-"), ".txt", sep=""),weighted = TRUE,threshold = 0.35,nodeNames = modProbes,altNodeNames = modGenes,nodeAttr = moduleColors[inModule])后记
本文使用的数据和参考文献的链接如下:
GEO Accession viewer (nih.gov)![]() https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE80565http://science.sciencemag.org/content/354/6312/aag1550
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE80565http://science.sciencemag.org/content/354/6312/aag1550![]() http://science.sciencemag.org/content/354/6312/aag1550
http://science.sciencemag.org/content/354/6312/aag1550
补充:
没有表型数据如何进行WGCNA分析?
WGCNA分析是专门针对基因表达谱数据的共表达网络分析方法,需要输入基因表达矩阵和与之对应的表型数据才能进行。表型数据包括每个样本的性状信息,例如疾病状态、年龄、性别等。
如果没有表型数据,可以考虑以下两种方法:
1. 使用外部数据合并:如果在其他研究中已经有合适的表型数据和相似的基因表达谱数据,可以将其合并,然后进行WGCNA分析。这种方法可以增加分析的样本数量,提高分析结果的可靠性和准确性。
2. 聚类分析:如果没有表型数据,也可以先进行聚类分析,将相似的样本归为同一组,然后对这些组进行WGCNA分析。这种方法需要注意聚类结果的可靠性和合理性,避免样本误分类导致结果不准确。
总之,无论采用哪种方法,都需要谨慎地处理数据,保证分析结果的可靠性和准确性。
后续会讲述如何使用Cytoscape软件进行网络可视化。
2023.8.30
----CXGG
这篇关于RNA-seq分析:Step9(共表达分析)的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!







