本文主要是介绍空间分辨率转录组学数据中的归因方法的性能比较评估,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
文章标题
A comparative performance evaluation of imputation methods in spatially resolved transcriptomics data
中文标题
空间分辨率转录组学数据中的归因方法的性能比较评估
文章链接
https://doi.org/10.1039/d2mo00266c
概况
之前介绍过一篇介绍sc-RNA-seq数据的各类插补方法,针对空间转录组的插补方法目前还是比较少,但是到现在为止,还是有一些基于图神经网络的方法出现的。本文评估了SpaGE、stPlus、gimVI、Tangram和stLearn五种插补的方法,通过各种评价指标与实验数据得出,目前已有的各类插补方法在空间转录组学的dropout问题上还是有所欠缺。
目前的空间转录组学手段:
1.基于原位杂交(situ hybridization):
smFISH、osmFISH、 MERFISH、 seqFISH+ 和 SRTARmap
2.基于原位捕获(situ capturing):
空间转录组学(ST/Visium),Slide-seq,HDST
数据
拥有同一PDAC病症的SRT数据,相应的图像和scRNA-seq数据的数据集从NCBI GEO数据库
https://www.ncbi.nlm.nih.gov/geo/
ID: GSE111672
健康的肝脏组织
ID: GSE185477
Visium空间转录组学数据集与同一研究中来自Donor2的scRNA-seq配对,其性别和年龄间隔相匹配。29个PDAC-A、PDAC-B和肝脏样本的SRT数据集分别有428、224和2231个spot(测序点)。
仿真数据
来源于一个sanger实验室的github:
模拟的SRT数据集有3000个点,是通过对小鼠大脑scRNA-seq数据中的43个参考细胞类型进行采样产生的,该数据集是由Kleshchevnikov等人制作的。合成数据(在本研究中被称为 “模拟ST”)的产生,考虑到了细胞丰度和数据集的稀疏性
https://github.com/emdann/ST_simulation/
验证手段
对已知的空间转录组数据,SpaGE、stPlus、gimVI和Tangram 会随机选择400个gene,从SRT数据集中删除 然后对被删除的基因的进行imputation。而stLearn不能对被剔除的基因进行预测,每个基因的5%(5stLearn)和30%(30stLearn)的非零值被随机选择并屏蔽为零,以便用stLearn进行归因。
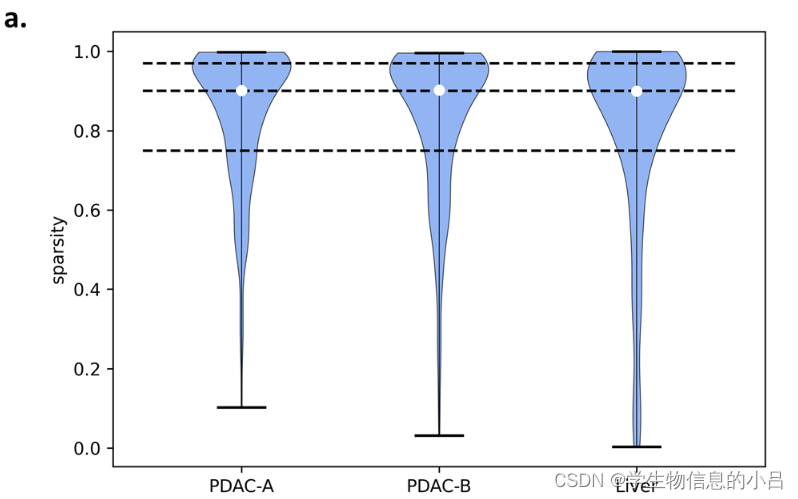
数据稀疏度图:
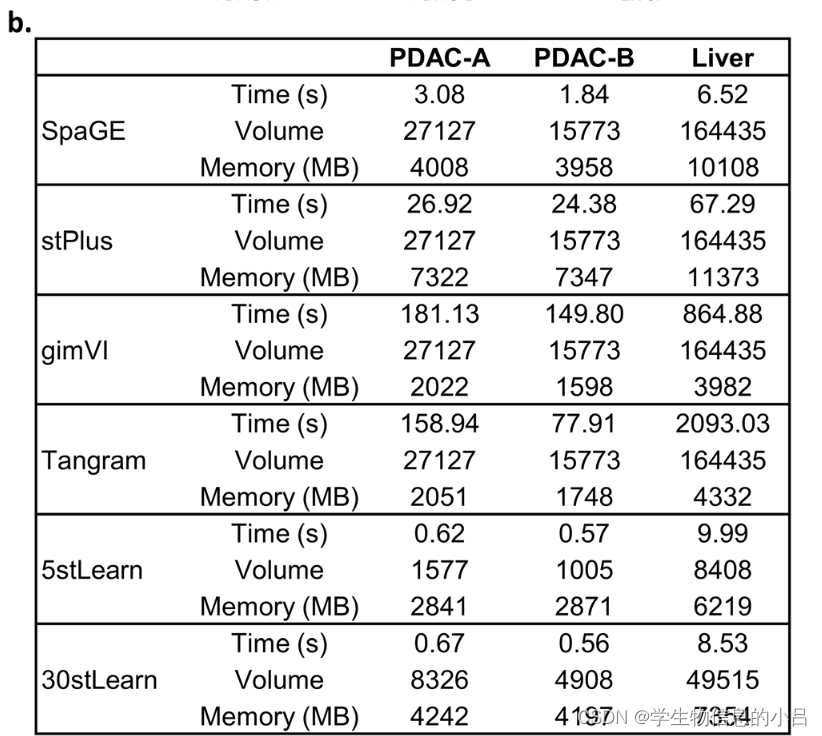
不同数据集和不同算法的计算开销
本文定义了 稀疏度 这一指标 即每种基因的计数值为0在整个spot集合中的比例。按照比例定义为:低稀疏、中度稀疏、高稀疏和非常高的稀疏。稀疏程度的边界是通过检查所有数据集中基因的稀疏程度分布来确定的。从每一组中选择100个gene。实则 仔细考虑这个办法是很有问题的。因为空间转录组数据本身到底什么spot位置 哪些基因会重点表达 本身是有内在联系的 但是随机武断的选择gene可能会掩盖某种方法在某种数据的真实性能发挥。。。
评价体系
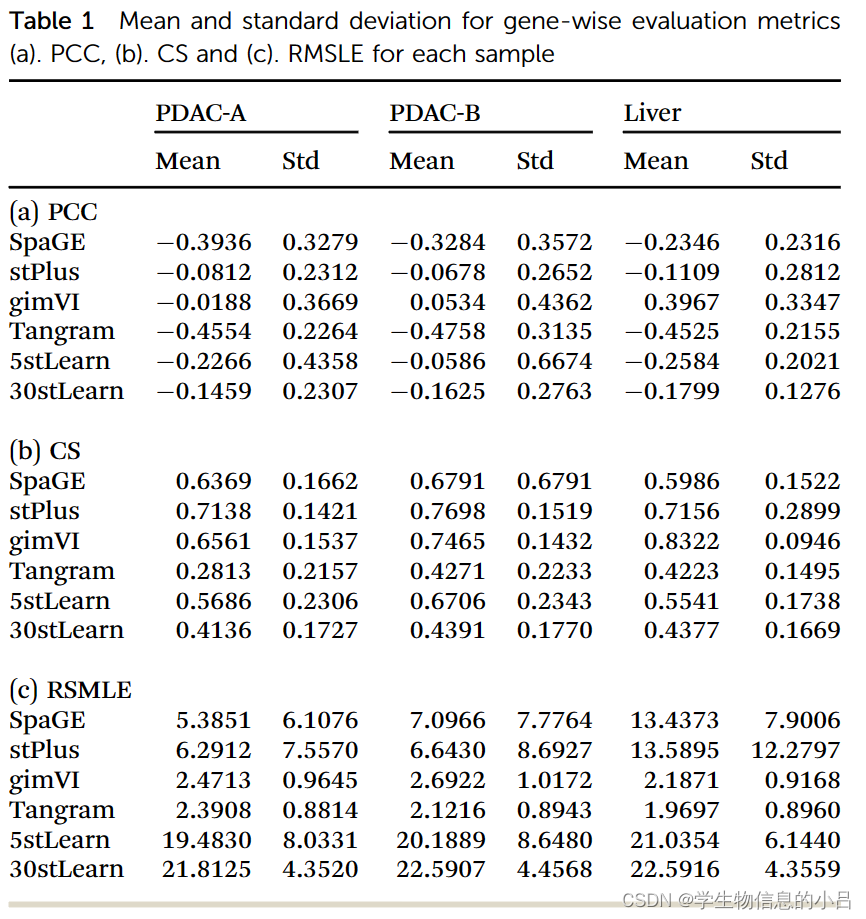
1.使用了皮尔逊相关系数(PCC)(这个不介绍了)、余弦相似度(CS)和均方根对数误差(RMSLE)等方法
余弦相似度介绍:

余弦相似度介绍
均方根对数误差(RMSLE)

RMSLE本身是在RMSE的前提下对数化,避免极大值的干扰。

2.使用Silhouette Index (SI) and Calinski Harabasz Index (CH)两个指标来评价聚类相似度。
这两个指标介绍了多次,这里不再介绍了。
结果
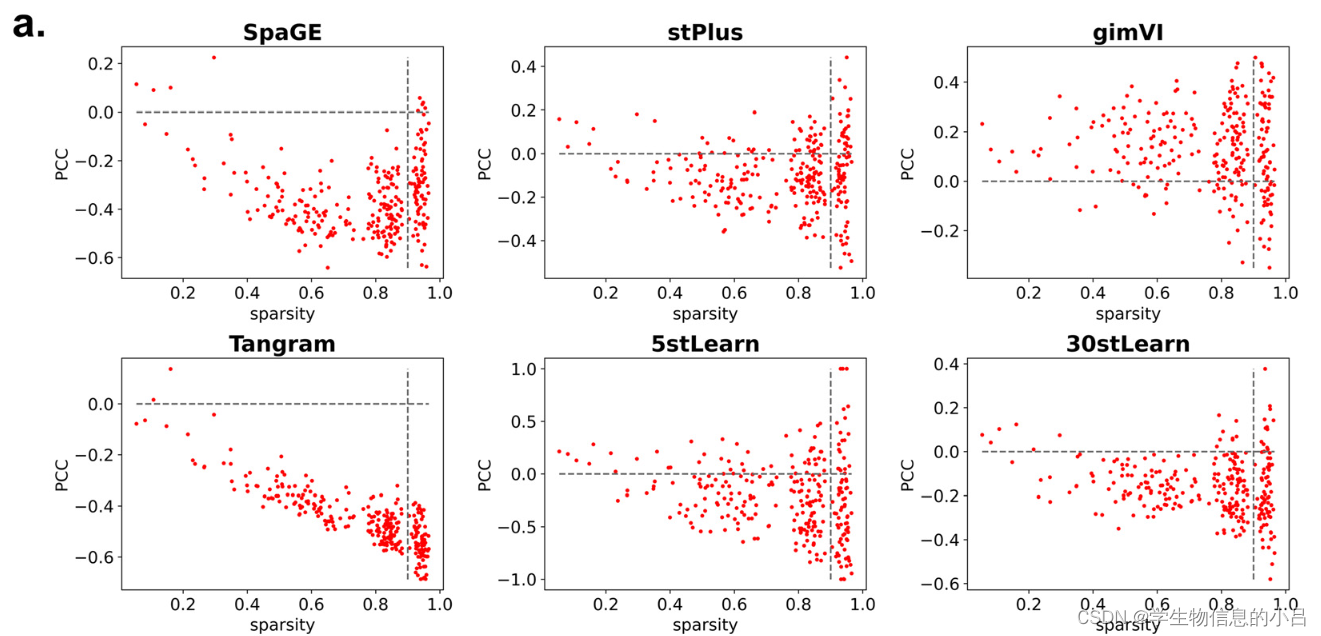
PCC指标中,gimVI是表现最佳的 ,但是
PCC本身遵从双变量正态分布 这个数学规律一般在gene上很难遇到,此外对偏度和极端的离散值很敏感也就是说 PCC这个指标能表达的含义有限,尤其是在空间转录组这种zero-rate超高的数据中,且空间转录组数据集本身有其自身平台特异性。
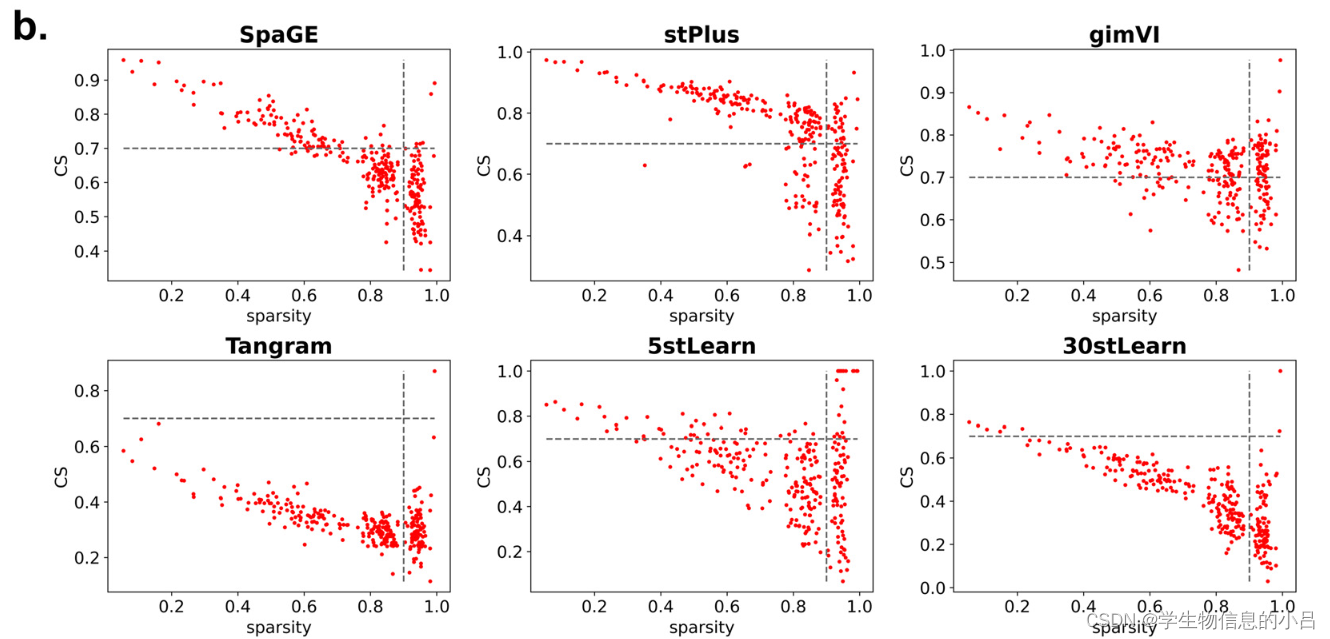
CS指标中,.高于0.7的CS被认为代表高相似度。平均CS值(stPlus为0.7330,gimVI为0.7449)和相似度图显示,stPlus和gimVI的预测结果分别有60.25%和72.00%的预测具有高相似度。与PCC值相比,相似性值意外地高得多。因此,为了评估CS值的显著性,进行了置换试验。结果显示,只有少数的相似性具有统计学意义(Po 0.05和CS Z 0.7)(图S2,ESI†)。
预测的误差用每个基因的RMSLE进行量化
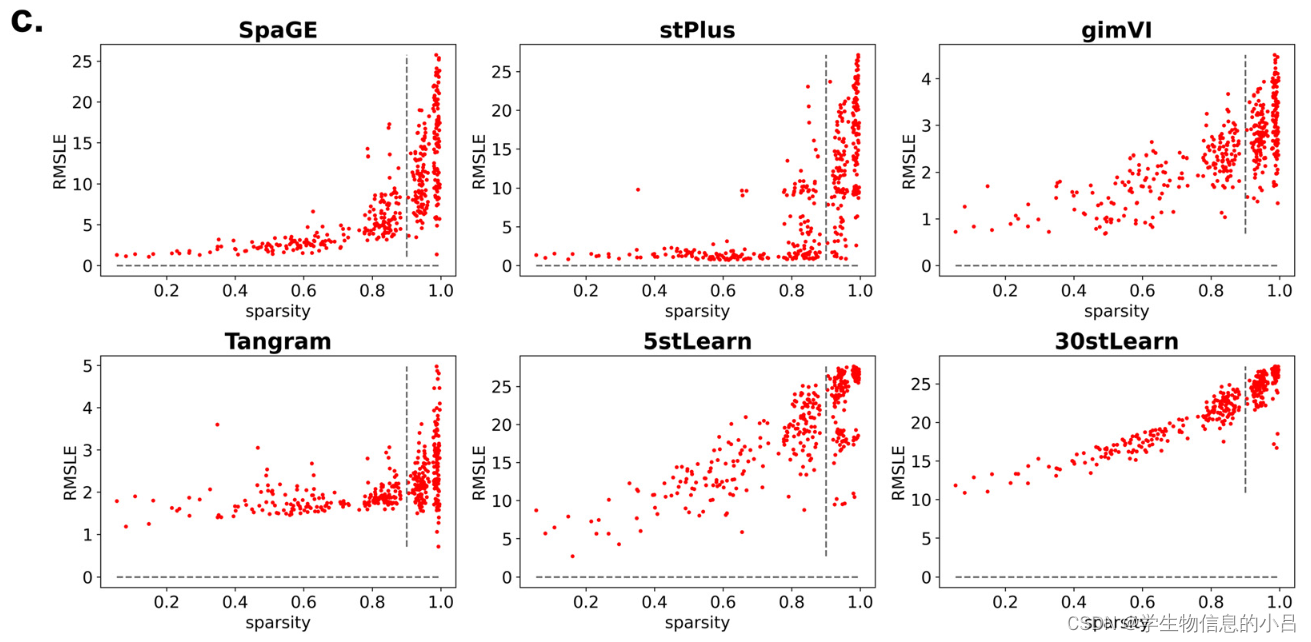
Tangram和gimVI的误差大多为零,表现优于其他公司。在RMSLE临界值为1的情况下,可以看到只有8.75%的stPlus和5.25%的gimVI的结果低于临界值。在定量评价指标方面,gimVI的所有指标都优于其他方法,而stPlus和Tangram在CS和RMSLE指标方面的表现与gimVI相当。
不同稀疏度基因集合的imputation效果
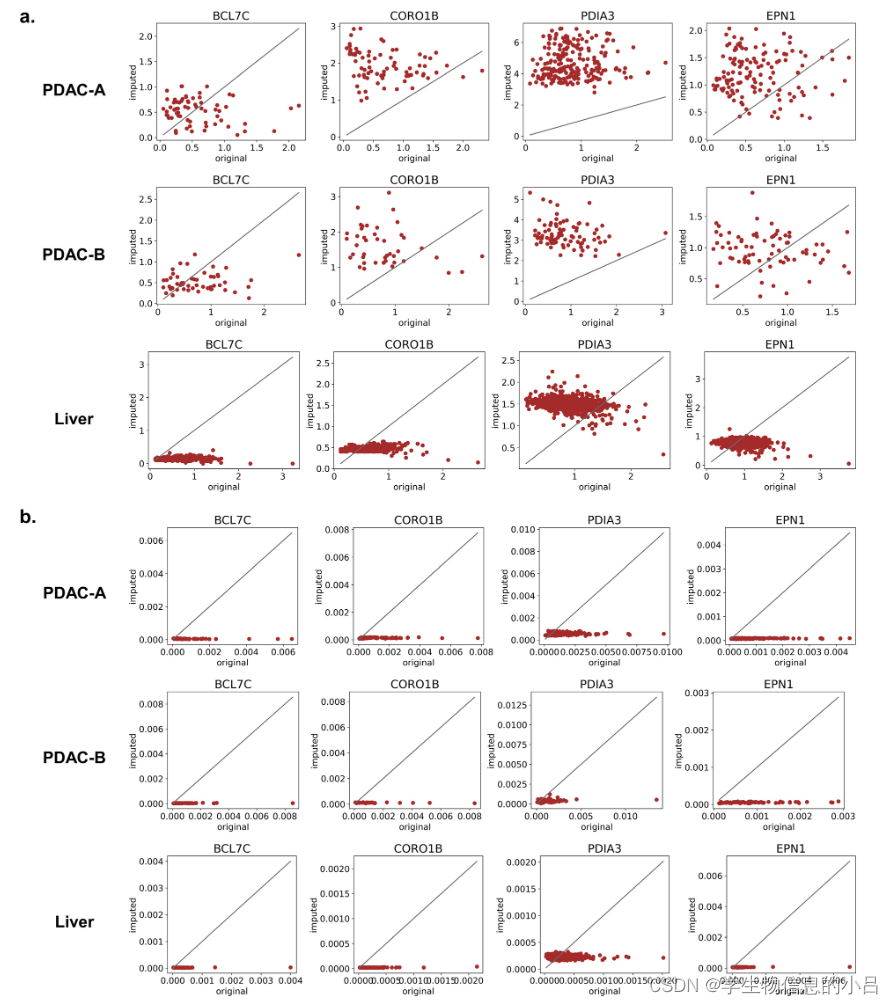
PDAC-A样本的原始非零计数及其预测值的可视化 每一组从左到右 是 不同稀疏度的数据集 在不同方法上的表现。(a) SpaGE, (b) stPlus, © gimVI, (d) Tangram, (e) 5stLearn和(f) 30stLean。
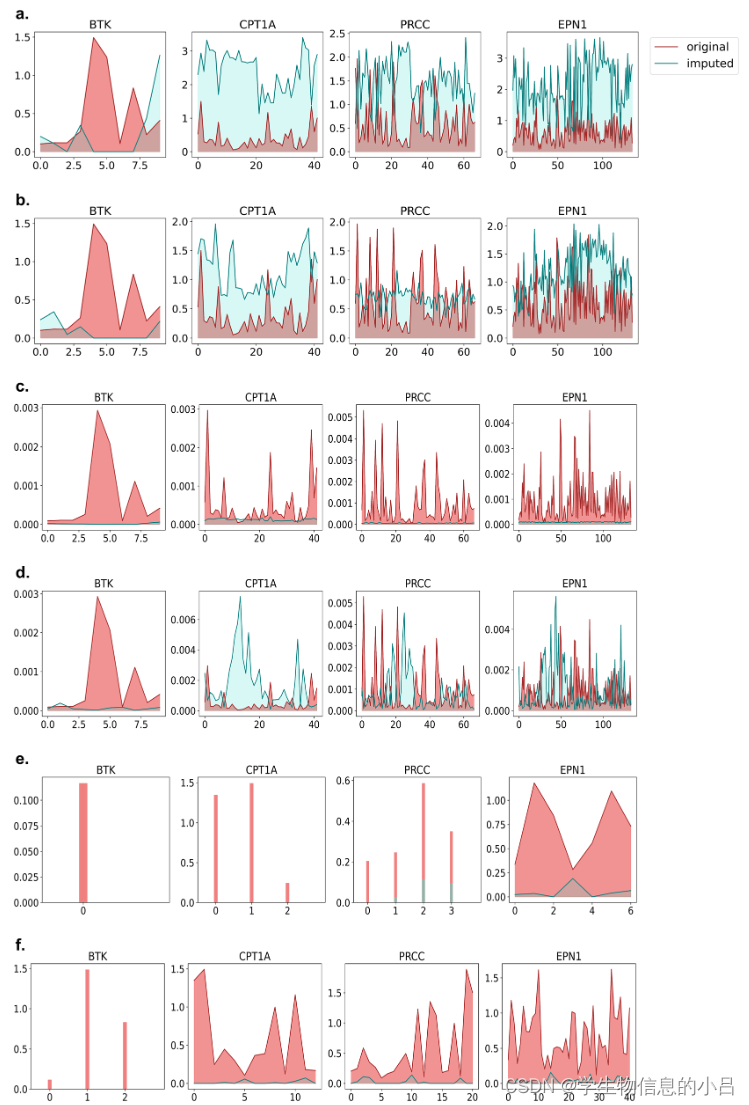
用每组随机选取的基因进行原始非零和相关预测的可视化。每一栏从左到右分别代表从非常高的稀疏度到低的稀疏度的基因组。发现预测结果大多接近于零,特别是对于具有非常高稀疏度的基因。此外,除gimVI和Tangram外,还观察到对原本不为零的计数值的零预测。
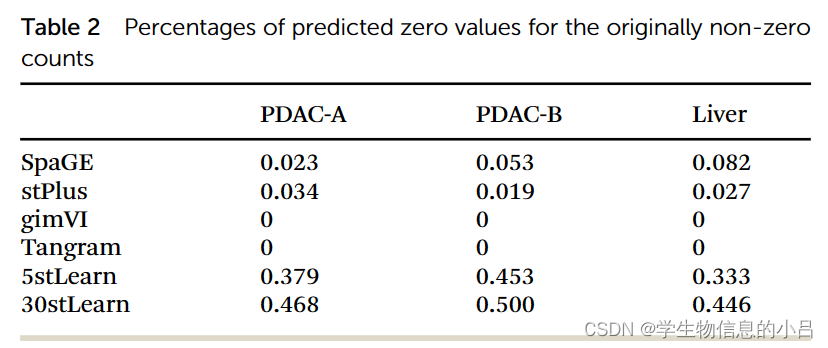
在SpaGE和stPlus中,零预测的百分比在1%到12%之间,而在stLearn中则在32%到50%之间。随着基因稀疏度的降低,零预测的百分比也在下降。
SpaGE和stPlus的预测值大多高于原始值,而与SpaGE对高稀疏、中度稀疏和低稀疏基因的预测相比,stPlus的预测值更接近原始值。gimVI的预测值低于原始值,在所有组中都接近于零。Tangram的预测值是可变的,有些预测值比原始值高,有些则比原始值低。
随着样本量的增加,SpaGE和Tangram的性能被发现是不变的,因为在样本量较低的样本中,性能指标是相似的
stPlus和gimVI的性能随着样本量的增加和基因稀疏度的降低而提高。在预测结果为零或接近零的情况下,增加样本量没有观察到stLearn的性能差异。
最后,发现stPlus和gimVI能够对样本量较高的低稀疏基因做出最佳估计。
stLearn的预测值比每组基因的原始值要小得多,这导致它与其他工具相比具有较低的性能。
从中度稀疏和低稀疏度的基因中选取 进行可视化:
辍学问题导致了基因表达谱中非常高和高的稀疏度,所以与其他稀疏度组的基因相比,具有这些稀疏度水平的基因被认为更需要归因。导致辍学的技术和生物问题也可能导致这些基因的非零计数的错误。然而,可以假设具有低或中度稀疏性的基因在数据集中被正确计数。因此,对这些基因的预测可以显示出对这些方法性能的更真实的看法。stPlus和gimVI被观察到在定量和视觉检查阻抗性能方面优于其他方法。因此,对这两种方法的中度和低度稀疏的基因进行了更详细的结果分析。从这两组中各随机选择两个基因进行可视化分析
观察到gimVI进行的预测大多低于中度稀疏的基因(BCL7C和CORO1B)的原始值。在低稀疏基因(PDIA3和EPN1)中,gimVI的预测结果与原始值比较接近,但其值仍然比原始值低得多,而且预测结果靠近原点。另一方面,stPlus的预测结果因样本大小而不同。在规模较小的样本中,观察到中度稀疏基因(BCL7C和CORO1B)的预测值低于或高于原始值,而低度稀疏基因(PDIA3和EPN1)的预测值则高于原始计数。由于样本量的增加,stPlus的预测得到了明显的改善。即使预测被定位在一个狭窄的区间内,即使对于中度稀疏的基因,也能获得与原始计数相同或接近的数值。
对随机选择的200个基因(来自中度和低度稀疏组)上的每个spot的基因表达谱进行评估,以揭示插补后spot特征的保存情况。检测了SRT数据集中每个斑点最初的非零计数,并计算了这些非零计数与这些中度和低度稀疏基因的相关预测值之间的 PC、CS和RMSLE。 高相关性、高相似性和低对数误差被认为是表明原始斑点特征的保存
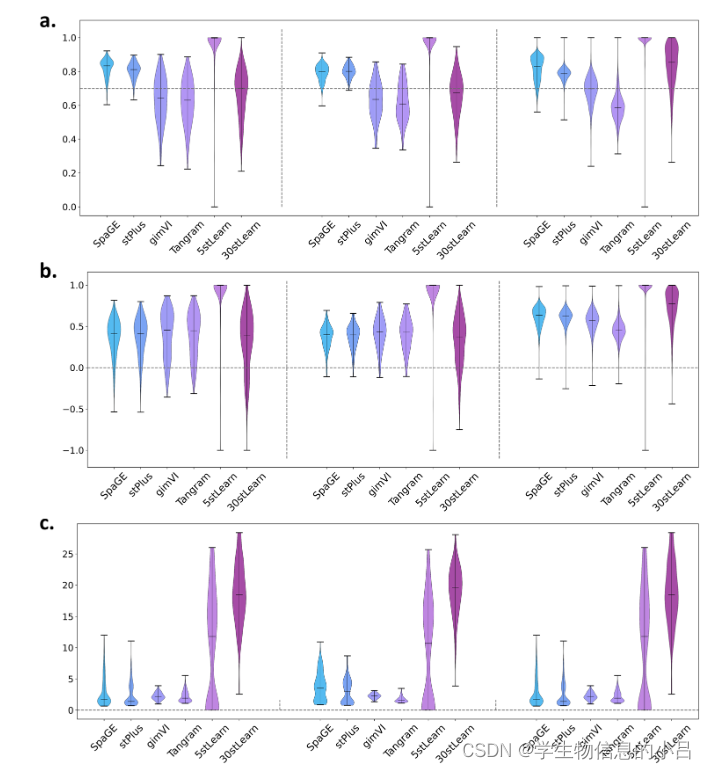
使用模拟数据集SRT
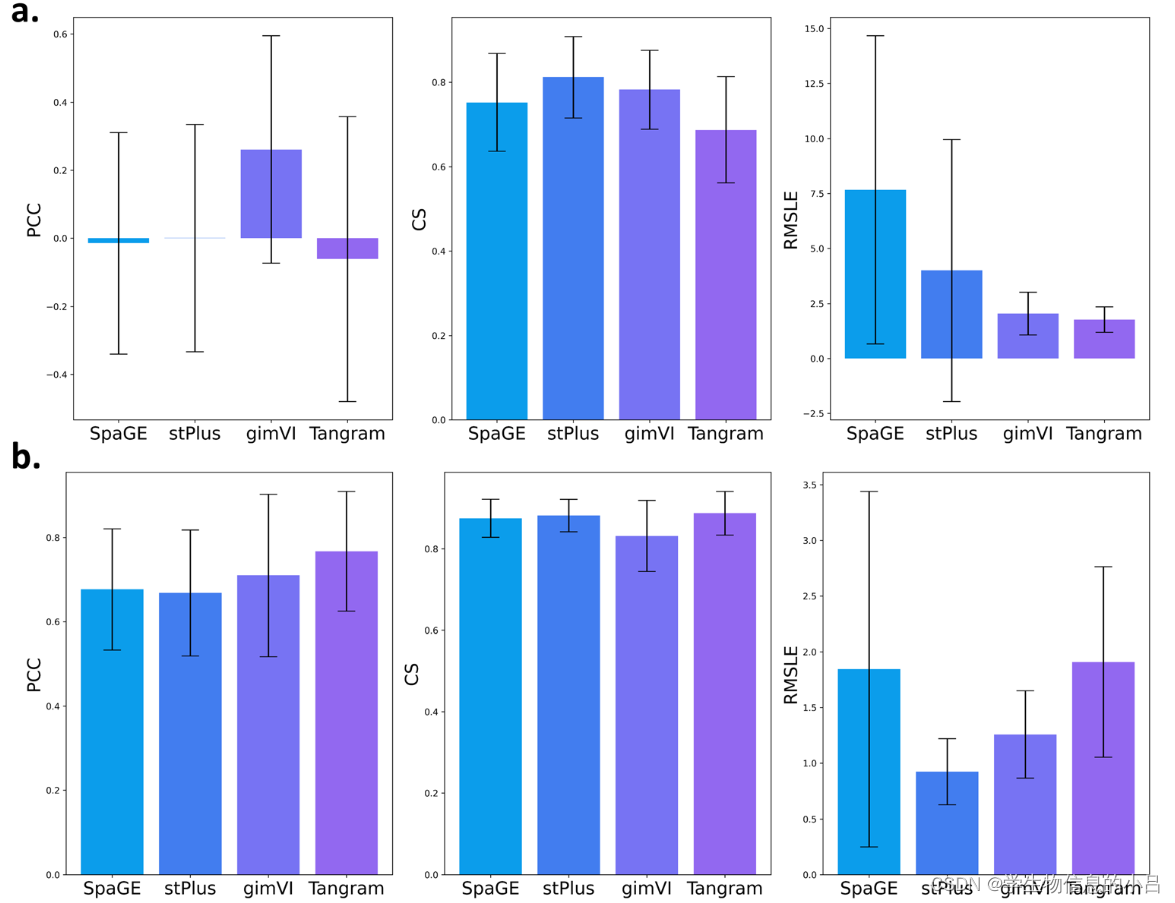
除了stLearn,其他四种方法都是基于参考的方法,以scRNA-seq数据集为参考。除了scRNA-seq中的辍学问题带来的偏差外,调查SRT数据集的匹配scRNA-seq并不总是可能的。此外,由于scRNA-seq和SRT数据集不是从完全相同的组织切片中产生的,因此它们之间总会有批次效应的变化。上述问题可能对基于参考的方法的性能有重大影响。因此,我们尝试从scRNA-seq生成一个合成的SRT数据集,并用这对数据集来评估这些方法,以消除缺乏来自匹配组织的实验数据的问题。与三个实验数据集相比,这四种方法在模拟SRT中的表现预计会更高。从小鼠脑部scRNA-seq产生模拟SRT数据集后,对之前确定的400个基因进行了归因处理。计算了基因和斑点的PCC、CS和RMSLE指标。
插补后数据进行聚类,计算CH和SI
Tangram在其他方法中表现出最好的聚类性能,具有最高的SI和CH。用Tangram对子空间上的聚类进行可视化处理更有优势,它消除了聚类图中的点的重叠。
总而言之
简而言之,具有 高稀疏度 的基因可以被认为是需要进行归纳的基因。相反,具有 中度和低度稀疏性 的基因被认为是可能 没有遇到辍学问题的基因 。基于这些假设,根据基因的稀疏程度对基因进行分组,并通过考虑基因的稀疏程度对这些方法进行详细评估是一种科学合理的方法。
上述结果中,PCC值大多为负值且接近于零,这些结果可以解释为在最初的非零值和相应的预测值之间没有发现任何关联性。CS值比PCC值高,但这些值在统计学上是不重要的。此外,最多有3%的计算误差被发现低于0.01。这些结果表明,在文献中,可靠的空间数据集估算方法仍然存在巨大的差距。我们希望这项工作将成为选择处理在整个转录组水平上产生的空间数据集的辍学事件的方法的指南。
这篇关于空间分辨率转录组学数据中的归因方法的性能比较评估的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!



