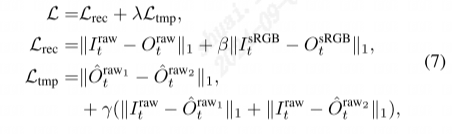本文主要是介绍贴梗海棠T2T基因组-文献精读40,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
A telomere-to-telomere reference genome provides genetic insight into the pentacyclic triterpenoid biosynthesis in Chaenomeles speciosa
端粒到端粒参考基因组为贴梗海棠中五环三萜生物合成提供了遗传学见解

摘要
贴梗海棠(Chaenomeles speciosa,2n=34)是一种蔷薇科的药用和食用植物,常用于传统中药中。然而,至今为止,缺乏基因组序列和遗传学研究阻碍了提高其药用价值的努力。在此,我们报告了一种整合性的研究方法,该方法结合了PacBio HiFi(三代测序)和Hi-C支架技术,组装了高质量的贴梗海棠端粒到端粒基因组。该基因组包含650.4 Mb,contig N50为35.5 Mb。其中,632.3 Mb被锚定在17条拟染色体上,其中12条拟染色体由单个contig表示,4条由两个contig表示,1条由四个contig表示。11条拟染色体在两端都有端粒重复序列,4条在一端有端粒重复序列。重复序列占基因组的49.5%,共注释了45,515个蛋白质编码基因。贴梗海棠的基因组大小与苹果(Malus domestica)相对相似。扩展或收缩的基因家族被识别并研究了它们与不同植物代谢或生物过程的关联。特别是功能注释表征了与齐墩果酸和熊果酸(贴梗海棠果实中两种丰富的五环三萜)生物合成途径相关的基因家族。总的来说,这个端粒到端粒、染色体级别的贴梗海棠基因组不仅为深入理解贴梗海棠组织中的药用化合物生物合成提供了宝贵资源,也促进了对蔷薇科植物进化的理解。
引言
贴梗海棠(Chaenomeles speciosa (Sweet) Nakai),又名木瓜或木瓜果,是一种属于蔷薇科的落叶灌木。它是一种二倍体物种(2n=34),原产于中国,并在全球广泛种植。木瓜海棠属包括五个物种:西藏木瓜海棠(C. thibetica Yü)、日本海棠(C. japonica (Thunb.) Lindl. ex Spach)、木瓜海棠(C. cathayensis (Hemsl.) Schneider)、贴梗海棠(C. speciosa (Sweet) Nakai)和木瓜(C. sinensis (Thouin) Koehne)。其中,贴梗海棠是一种可食用的药用植物。它的果实具有酸味,是传统中药中使用超过千年的几个重要植物部位之一。其干果被认为具有平肝、舒筋活络、和胃、祛湿的功效。在中医药中,其干果的药用包括治疗哮喘、肝炎、消化不良、痢疾、肠炎和类风湿性关节炎。此外,它的果实还广泛用于制作果酱和果冻。
迄今为止,贴梗海棠干果的药用促使了多项研究以了解其活性成分。植物化学分析表明,贴梗海棠的果实含有三萜类化合物、酚类和酚丙烷酸类、黄酮类、糖类和生物碱。在这些三萜类化合物中,中国药典指定齐墩果酸和熊果酸是贴梗海棠的两个特征性代谢标志物。最近的代谢组学分析进一步揭示其果实富含对健康有益的代谢物,具有药用价值,如抗炎、镇痛、抗菌、抗病毒、抗氧化、抗流感和免疫调节活性。这些发现表明,进一步研究贴梗海棠药用价值背后的植物化学基础具有重要意义。鉴于目前对这种药用植物的植物代谢了解有限,需要通过基因组学、活性植物化学物质的生物合成阐明、代谢工程和遗传学等研究来成功生产活性化合物。
蔷薇科是一个包含超过3000个物种的大科,其中苹果、桃子、梨子、李子、草莓、樱桃、覆盆子和玫瑰是常见的经济作物,有益于人类健康。因此,蔷薇科适合用于研究水果多样性、驯化和进化。目前,已经对几种物种的基因组进行了测序,显示出在增强遗传学理解、育种和其他研究工作中的价值。基于下一代测序技术生成的短读长,这些测序研究为相关植物提供了基因组注释流程。此外,尽管存在某些技术限制,这些完整、准确且连续的代表性基因组序列为其他物种的进一步基因组分析提供了有价值的参考。更重要的是,这些可用的基因组序列对于木瓜属物种的基因组注释也是有用的参考,迄今为止,据我们所知,尚未对任何木瓜属物种进行测序。
目前,在长读长测序技术和组装流程方面已经取得了进展。这些新技术,如高保真(HiFi)测序,作为一种第三代方法,已用于生成多个染色体级基因组组装的基因组序列,具有更高的完整性、连续性和准确性。更重要的是,HiFi测序已被用于开发作物的端粒到端粒(T2T)和无缺口参考基因组,并显示出在识别重复区域中的独特基因和结构变异方面的能力。因此,我们报告了使用这一新技术对异二倍体贴梗海棠的基因组进行测序和组装。生成的T2T基因组质量高,为了解贴梗海棠中三萜类化合物的生物合成提供了有用信息。此外,这些基因组序列为理解驯化对序列变异的影响以及蔷薇科植物的进化提供了宝贵资源。
结果
贴梗海棠基因组的全新组装
我们结合短读长、HiFi和Hi-C测序对贴梗海棠的基因组进行了测序和组装。首先,我们完成了短读长测序,获得了660 Gb的双端读数。K-mer分析结果(k=21)显示基因组大小约为650.40 Mb,异质性率为2.1%,并且我们在~50和100的深度下分别识别出一个异质峰和一个同质峰。详细分析表明,基因组包含约257.0 Mb的重复序列。接下来,我们使用高质量的DNA样本构建了一个SMRTbell文库,并使用PacBio Sequel II系统对其进行了测序。此测序生成了共计18.8 Gb的HiFi读长,N50长度为19.0 kb(28.92×覆盖率)。然后我们使用Hifiasm流程组装HiFi读长,获得了一个包含167个contig的初步组装,总长度为650.4 Mb。最长的contig长度为51.8 Mb,其中50%的contig长度超过35.5 Mb,19个contig覆盖了总长度的90%。

图1 贴梗海棠基因组的调查和组装。(A)绿色的果实,(B)两套贴梗海棠单倍体染色体的共线性。这种共线性基于两套染色体之间的端粒、重复序列分布和基因密度。(C)贴梗海棠的高质量基因组组装。基因组图谱包含五个环圈。最外层的绿色环圈由17条染色体组成。其余的环圈分别表示染色体上基因、转座子、端粒重复序列和GC含量的分布。中心代表染色体之间的共线性区块。(D)展示k-mer分析产生的异质性图。(E)展示苹果和贴梗海棠基因组之间共线性相似性的图。
| Hap1 | Hap2 | |||||
|---|---|---|---|---|---|---|
| Contigs | Scaffolds | Pseudo-chromosomes | Contigs | Scaffolds | Pseudo-chromosomes | |
| No. of assembly | 230 | 125 | 17 | 97 | 42 | 17 |
| Total length (bp) | 605 875 599 | 605 928 099 | 592 846 119 | 632 365 991 | 632 393 491 | 608 284 049 |
| N50 (bp) | 25 567 090 | 36 115 581 | 36 115 581 | 29 342 999 | 36 916 915 | 36 916 915 |
| L50 | 10 | 7 | 7 | 8 | 7 | 7 |
| N90 (bp) | 5 854 963 | 29 924 437 | 29 924 437 | 8 796 568 | 30 504 571 | 30 700 526 |
| L90 | 30 | 14 | 14 | 25 | 14 | 15 |
| Gap Number | 0 | 103 | 61 | 0 | 45 | 26 |
| Max length (bp) | 38 639 417 | 51 774 635 | 51 774 635 | 40 832 825 | 52 302 129 | 52 302 129 |
| GC content (%) | 37.59 | 37.59 | 37.41 | 37.42 | 37.25 | 37.25 |
| No. of protein-coding genes | 40 874 | 43 836 | ||||
| BUSCO (genome) | C: 96.2% (S: 66.4%, D: 29.8%), F: 0.8%, M: 3.0%, n:1440 | C: 96.2% (S: 64.3%, D: 31.9%), F: 1.3%, M: 2.5%, n:1440 | ||||
| BUSCO (protein-coding genes) | C: 90.1% [S: 64.5%, D: 25.6%], F: 5.3%, M: 4.6%, n:1440 | C: 91.6% [S: 64.2%, D: 27.4%], F: 4.9%, M: 3.5%, n:1440 |
为了锚定这些contig,从Hi-C文库生成了总计40.0 Gb的双端读长。经过使用HiCUP清理和过滤读长后,结果显示94.2%为有效对。所有有效对都被用于支架组装,最终获得了122个支架,其中16个支架覆盖了总长度的90%(表2)。基于Hi-C测序数据,将25个支架锚定到17条拟染色体上(图S1,见在线补充材料)。该染色体级别的基因组组装包含632.3 Mb的序列,支架N50长度为35.8 Mb,GC含量为37.3%(表2)。进一步的序列分析表明,17条拟染色体的长度范围从24.5 Mb到51.8 Mb不等。在这17条拟染色体中,有12条由一个大contig组成,4条由两个contig组成,1条由四个contig组成(表S1,见在线补充材料)。对拟染色体末端的序列分析显示,11条染色体在两端都有端粒重复序列,4条染色体在一端有端粒重复序列(图1C;表S1,见在线补充材料)。染色体LG01和LG06缺乏端粒重复序列。使用Centromics程序鉴定着丝粒,结果表明每条染色体上都有着丝粒(图S2,表S2,见在线补充材料)。此外,在组装的17条染色体中发现了七个间隙(表S3,见在线补充材料)。为了表征间隙两侧各20 kb(上游10 kb和下游10 kb)区域的序列,使用MUMmer程序进行了共线性分析。结果数据显示在这些间隙两侧20 kb区域中有大量的分散重复和反向重复,例如在染色体8上的间隙(LG08Gap1::LG08:16430026–16 450 026)的11 kb至约20 kb为反向重复,这一区域在染色体9上第三个间隙(LG09Gap3::LG09:26623338–26 643 338)的0 kb至9 kb位置也有一个复制(分散重复)(图S3,见在线补充材料)。此外,GC含量分析显示间隙两侧序列的GC含量范围为35.16%至39.92%,平均值为37.54%。17条染色体中的GC含量范围为36.29%至37.94%,平均值为37.31%。Student’s t检验分析显示间隙两侧序列与整个染色体的GC含量之间没有显著差异(P=0.7391)。此外,我们在七个间隙两侧序列中提取了10 kb长的核苷酸,获得了八个基因,这些基因被注释为一个转录因子、受体和其他(表S4,见在线补充材料)。
| * C. speciosa* genome | |||
|---|---|---|---|
| Contigs | Scaffolds | Pseudo-chromosomes | |
| No. of assembly | 167 | 122 | 17 |
| Total length (bp) | 650 391 965 | 650 410 993 | 632 305 965 |
| N50 (bp) | 35 454 787 | 35 780 769 | 35 780 769 |
| L50 | 8 | 8 | 8 |
| N90 (bp) | 16 814 540 | 29 516 134 | 32 355 895 |
| L90 | 19 | 16 | 15 |
| Gap number | 0 | 45 | 7 |
| Max length (bp) | 51 822 334 | 51 822 334 | 51 822 334 |
| GC content (%) | 37.39 | 37.47 | 37.31 |
| No. of protein-coding genes | 45 515 | ||
| BUSCO (genome) | C: 96.5% (S: 65.0%, D: 31.5%), F: 1.0%, M: 2.5% | ||
| BUSCO (protein-coding genes) | C: 93.7% [S: 62.0%, D: 31.7%], F: 4.1%, M: 2.2% |
接下来,使用Illumina测序数据构建了一个Hi-C文库。HiFiasm结合HiFi数据提供了精确的局部单倍型信息,而Hi-C数据提供了长距离相互作用信息,从而实现了基因组的全局分型,鉴定出贴梗海棠基因组的两个单倍型,Hap1和Hap2。这两个单倍型包含34条染色体。Hap1的基因组大小为592.84 Mb,特征为230个contig(N50为25.57 Mb)、125个支架(N50为36.12 Mb)和37.41%的GC含量。Hap2的基因组大小为608.28 Mb,特征为97个contig(N50为29.34 Mb)、42个支架(N50为36.92 Mb)和37.25%的GC含量(表1;表S5和S6,见在线补充材料)。基于这些序列,在Hap1中,有8条染色体在两端有端粒重复序列,8条染色体在一端有端粒重复序列;在Hap2中,有10条染色体在两端有端粒重复序列,5条染色体在一端有端粒重复序列(图1B;表S7和S8,见在线补充材料)。通过基准统一单拷贝直系同源(BUSCO)分析进行的进一步质量评估显示,Hap1覆盖了胚乳植物_odb 10基因集的96.2%,Hap2的覆盖率也是96.2%(表2)。最后,基于Hap1和Hap2的基因组,我们组装了包含全部34条染色体的贴梗海棠基因组,并构建了一个圆形图谱来可视化其基因组(图1C)。
我们根据上述的组装对Hap1和Hap2的基因组进行了比较。数据表明这两个单倍型在基因组大小和基因数量方面具有相似性(表2)。全基因组比对进一步揭示了两个单倍型之间的大规模保守共线性(图1B)。我们还分析了两个单倍型之间的单核苷酸多态性(SNP)、插入缺失(InDels)和结构变异(SV)。数据揭示了Hap1和Hap2之间明显的差异,包括6,972,451个SNP、987,101个InDels、73个收缩、5461个插入、4587个缺失、70个倒位、125个易位和184个重复(图1B;图S4,表S9-S11,见在线补充材料)。这些数据提供了大量的信息,帮助理解两个单倍型之间的遗传变异。序列表征显示,258,544个SNP和InDels(包括876个起始密码子、6614个终止密码子和10,597个剪接位点、错义变体和移码)有助于贴梗海棠基因功能的多样性(约2.7%的品种)。
从Hap1和Hap2基因组推导出的氨基酸序列被用于聚类分析(mcl程序,参数-I 2.0 -abc)。结果数据返回了28,555个基因家族。其中,10,487个基因家族来自于26,966个Hap1基因和27,923个Hap2基因的注释,并被表征为多直系同源基因家族。此外,11,382个基因家族被表征为单直系同源基因家族,其中2395个基因家族来自2526个单基因,被注释为Hap1特异性,4291个基因家族来自4531个单基因,被注释为Hap2特异性(表S12-S14,见在线补充材料)。此外,我们使用京都基因与基因组百科全书(KEGG)、基因本体(GO)和蛋白质家族分析与建模(PFAM)进行了富集分析,以表征Hap1和Hap2特异性基因。KEGG结果显示,Hap1特异性基因在氨基酸生物合成(ko01230)、甲状腺激素信号通路(ko04919)和植物-病原体相互作用通路(ko04626)中富集,而Hap2特异性基因在剪接体(ko03040)、甜菜碱生物合成(ko00965)和维生素B6代谢(ko00750)中富集(Fisher精确检验,P < 0.05)(图S5和S6,见在线补充材料)。GO结果显示,Hap1特异性基因在DNA整合(GO: 0015074)、细胞对刺激的反应(GO: 0051716)和信号传导功能(GO: 0007165)中富集,而Hap2特异性基因在DNA整合(GO: 0015074)、细胞周期(GO: 0007049)和DNA代谢过程功能(GO: 0006259)中富集(Fisher精确检验,P < 0.05)(图S7-S12,见在线补充材料)。PFAM结果表明,Hap1和Hap2特异性基因都在逆转录酶(RNA依赖的DNA聚合酶)(RVT_2)、整合酶核心结构域(rve)和逆转录酶中的锌结合(zf-RVT)中富集(图S13和S14,见在线补充材料)。
基因组质量评估
为了评估组装质量,我们完成了BUSCO分析。结果数据表明,在组装的基因组中,96.5%的BUSCO存在(表2)。单拷贝和重复基因分别占组装基因组的65.0%和31.5%。这些数据表明贴梗海棠基因组具有高度的全基因组重复性。BUSCO分析还表明仅有2.5%的缺失基因和1%的缺失片段,表明基因组组装几乎是完整的。
长末端重复序列(LTR)组装指数(LAI)评分、读数比对和拟染色体比较是评估基因组组装质量的另外三种重要方法。首先,LTR预测显示LAI评分为11.23。这个高分数表明基因组组装的高质量。同时,用于基因组调查的双端读长与组装进行了比对,结果显示高比对率达到98.02%。第二代测序调查数据对基因组的平均覆盖深度为106.33倍;开发了数据的深度分布图,以可视化每条染色体的二代测序序列质量(图S15,见在线补充材料)。最后,我们将我们的序列与二代测序获得的序列进行了比较,并使用GATK 4.0进行了SNP和InDel分析。此比较从17条染色体中获得了4386个(每条染色体平均258个)纯合SNP和InDel。此外,这次比较显示整个染色体大小的SNP和InDel比例为6.936515e−06,减少到5.021475e−06到8.560801e−06,表明每条染色体的比例值。进一步的统计分析表明,组装染色体的单个碱基准确率超过99.999%。(表S15和图S16,见在线补充材料)。这些统计数据表明组装的高准确性。
此外,将贴梗海棠的拟染色体与苹果的拟染色体进行了比较,以了解任何结构和序列的相似性。两个基因组在染色体水平上显示出紧密的一对一共线性关系(图1E,表S16)。使用MCScanX的分析检测到231个共线性区块(表S17,见在线补充材料),这些区块平均包含375个基因。最大的区块包含2670个基因,最小的包含33个基因。这些区块覆盖了贴梗海棠的26,391个基因(占总基因的57.98%)和苹果的25,148个基因(占总基因的57.98%)。此外,贴梗海棠和苹果之间的蛋白质序列用Blast(阈值:e值<1e-5,最小覆盖率>40%)进行了比较。结果数据显示,10,029个基因簇(34,352个贴梗海棠基因和34,991个苹果基因),其中9704个是贴梗海棠特有基因,而10,521个是苹果特有基
因。为了了解它们的潜在功能,我们对贴梗海棠特有基因进行了富集分析。KEGG的结果显示,贴梗海棠特有基因与凋亡(KO04210)、碳代谢(ko01200)以及间隙连接和其他代谢通路(Ko04540)有关(Fisher精确检验,P < 0.05)。保守的Pfam结构域分析结果显示,这些贴梗海棠特有基因的富集功能是MULE转座酶结构域、SWIM锌指蛋白和逆转录酶(P < 0.05)(表S18,见在线补充材料)。GO功能分析表明,这些基因在RNA-DNA杂交核糖核酸酶活性(GO: 0004523)、核酸内切酶活性(GO: 0016893)、核糖核酸内切酶活性(GO: 0004521)等功能中富集(表S18-S21,见在线补充材料)(Fisher精确检验,P < 0.05)。综上所述,这些结果不仅表明了组装的贴梗海棠基因组的高连续性、完整性和准确性,还显示了该组装是一个有价值的参考基因组。
基因组元素的注释
对基因组元素进行注释以表征组装的基因组。在组装的基因组中使用de novo预测和基于同源性的搜索方法识别了重复序列,总共识别了312.9 Mb的重复序列,占基因组的49.5%。序列分析显示,转座元件(TEs)构成了重复序列的主要部分。RNA逆转座子(I类)是TEs的主要部分。在这些逆转座子中,LTR是最丰富的,占整个基因组的36.3%(表S22,见在线补充材料)。主要的LTR逆转座子是Gypsy元件(22.7%),其次是Copia元件(7.3%)。除I类外,DNA转座子(II类)占基因组的1.0%,其中PIF-Harbinger最为丰富。
使用ab initio、基于同源性搜索和Iso-seq数据的结合方法预测了基因模型。结果数据预测了45,515个高可信度的编码蛋白质基因(占组装的19.0%),平均长度为2636 bp。在贴梗海棠基因组中,所有这些编码基因都通过七个公共数据库进行了功能注释。结果显示,通过NCBI、NR数据库、TrEMBL数据库、Pfam数据库、SwissProt蛋白质数据库、KOG数据库、GO数据库和KEGG数据库分别注释了93%、90%、70%、67%、48%、43%和29%的编码基因。所有这些数据库允许对93%的编码基因进行功能注释。此外,基因模型获得了93.7%的BUSCO,包括62.0%的单拷贝基因和31.7%的重复基因(表2)。
此外,基因组组装还识别了4205个核糖体RNA(rRNAs)、685个转运RNA(tRNAs)、274个小核RNA(snRNAs)和739个小核仁RNA(snoRNA)。基于序列分析和注释,我们开发了一个Circos图(图1C),可视化了染色体上的基因、转座子、端粒重复序列和GC含量的分布以及染色体之间的共线性区块。此外,图中包含的共线性区块分析数据表征了基因组区域的序列相似性,这些区域通过线条连接。共线性区块分析进一步揭示了同源染色体对,如LG01和LG7、LG02和LG15、LG03和LG11、LG04和LG12、LG05和LG10、LG06和LG14、LG08和LG15、LG09和LG17、LG13和LG16,源自四倍化事件(图1C)。
贴梗海棠的直系同源聚类和系统发育地位
为了理解贴梗海棠基因组的进化,我们完成了贴梗海棠、葡萄(Vitis vinifera)、番木瓜(Carica papaya)、拟南芥(Arabidopsis thaliana)和蔷薇科七个代表物种(苹果(Malus domestica)、杏(Prunus armeniaca)、桃(Prunus persica)、樱花(Prunus yedoensis)、白桦梨(Pyrus betulifolia)、欧洲梨(Pyrus communis)和玫瑰(Rosa chinensis))的直系同源聚类。根据序列相似性,所有编码蛋白质的基因被聚类成6968个基因家族,这些基因家族在这11个物种中被普遍识别。与此同时,在贴梗海棠基因组中识别出了5658个特异性基因家族(图2A)。通过GO富集进行的生物过程分析表明,这些特异性基因主要参与辅因子代谢、辅酶代谢和甲硫氨酸代谢过程。通过GO进行的分子功能分析显示,它们参与了核酸内切酶活性、丙酮酸激酶活性和钾离子结合(图S17,见在线补充材料)。与此同时,KEGG富集分析表明,这些物种特异性基因主要参与代谢过程,包括碳代谢、丙酮酸代谢、半乳糖代谢和萜类骨架生物合成(图S18,见在线补充材料)。这些结果表明,贴梗海棠已经进化出了与初级和次级代谢物的生物合成相关的独特遗传和分子机制。

图2 贴梗海棠基因组的直系同源聚类和系统发育分析。(A)花形图展示了贴梗海棠和其他10个植物物种共享的核心直系同源组(位于中心)以及由11个物种特异性直系同源组形成的11个花瓣部分。(B)通过KEGG进行的富集分析表明,贴梗海棠基因组中扩展的基因家族与17种不同的功能相关。(C)从11个基因组构建的系统发育树显示了贴梗海棠和其他10个物种之间的进化血统关系。分支节点上的数字表示分歧时间。每个分支中的饼图显示了基因组大小扩展(右侧色块)和收缩(左侧色块)的相对比例。每个植物物种中扩展(+)和收缩(−)的基因家族数量标注在右侧。
贴梗海棠基因组的系统发育地位通过使用其他10个植物基因组进行分析(图2C)。该分析从11个植物基因组中鉴定出140个单拷贝直系同源组。使用这些11个植物物种的基因组和拟南芥(A. thaliana)作为外群物种,构建了一个跨基因组的系统发育树并估计了分歧时间。结果树显示,贴梗海棠和苹果(M. domestica)聚集在一起,并与白桦梨(P. betulifolia)和欧洲梨(P. communis)形成一个簇。在分类学上,这四个物种属于蔷薇亚科(Amygdaloideae)中的苹果族(Maleae),表明它们的基因组可能源自同一古代祖先。杏(P. armeniaca)、桃(P. persica)和樱花(P. yedoensis)属于蔷薇亚科中的桃族(Amygdaleae),它们聚集在一起。而玫瑰(R. chinensis),属于蔷薇亚科中的蔷薇族(Roseae),则与蔷薇亚科的其他物种分离。这些结果显示,这些植物在系统发育树中的拓扑位置与其植物系统发育一致。贴梗海棠和苹果的分歧时间发生在约1000万年前(MYA)。苹果族起源于约10.92 MYA的桃族祖先。蔷薇科物种的分歧发生在约98.81 MYA。
基因家族的扩展和收缩
为了理解基因组中谱系特异性动态变化,我们挖掘了注释数据以识别显著扩展或收缩的基因家族(图2C)。结果在贴梗海棠基因组中识别出158个收缩的基因家族和320个扩展的基因家族。这些数据表明,贴梗海棠和苹果基因组中扩展的基因家族数量相似。此外,与蔷薇科中的其他物种相比,贴梗海棠中扩展的家族数量较高(图S19和S20,见在线补充材料)。相比之下,贴梗海棠中收缩的基因家族数量约为苹果的一半。GO富集分析显示,收缩的基因家族参与了生物过程类别中的“脂质运输”和“信号传导”,以及分子功能类别中的“ADP结合”和“鲨烯单加氧酶活性”(图S21,见在线补充材料)。KEGG分析揭示,收缩的基因家族与氰基氨基酸代谢、类固醇生物合成、苯丙烷类生物合成、倍半萜类和三萜类生物合成、淀粉和蔗糖代谢、以及茄啶、胡椒碱和烟碱生物合成有关(图S22,见在线补充材料)。与此同时,KEGG富集分析表明,扩展的基因家族与单萜类、生物合成、白藜芦醇、生物合成、二芳基庚酮、生姜醇、生物合成、亚油酸、生物合成、黄酮类、生物合成、倍半萜类、生物合成、三萜类生物合成有关(图2B)。
转座元件的扩增
序列挖掘显示,转座元件(TEs)是贴梗海棠基因组大小扩展的一个主要事件。为了理解TEs在这种扩展中的作用,对葡萄(V. vinifera)、玫瑰(R. chinensis)、杏、桃、欧洲梨、贴梗海棠和苹果的TE含量进行了比较(图3A)。根据TE类型,基因组被分类为非重复、LTR重复和非LTR重复类型。结果显示,苹果和贴梗海棠的基因组在进化过程中具有相似的LTR重复和非LTR重复含量。相比之下,杏和桃的基因组中LTR重复和非LTR重复的含量有所减少。进一步对LTR的插入时间进行分析,表明LTR的增殖发生在杏和贴梗海棠约0.05和0.08 MYA之间,玫瑰、梨、苹果和桃的增殖发生在约0.15 MYA之间(图3B)。此分析还揭示,葡萄中的LTR插入时间比其他六个物种要早得多。

图3 贴梗海棠的全基因组重复(WGD)、共线性和染色体进化特征。(A)比较了葡萄(Vitis vinifera)、玫瑰(Rosa chinensis)、杏(Prunus armeniaca)、桃(Prunus persica)、欧洲梨(Pyrus communis)、贴梗海棠(C. speciosa)和苹果(Malus domestica)基因组中的重复序列含量。(B)比较了葡萄、玫瑰、杏、桃、欧洲梨、贴梗海棠和苹果中的LTR插入时间。(C)图表显示了贴梗海棠的自共线性分析结果特征。(D)使用蔷薇科六个物种构建了系统发育树。(E)从玫瑰、杏、桃、欧洲梨、贴梗海棠、苹果的跨基因组共线性分析中创建了一个图表。(F)图表显示了玫瑰、杏、桃、欧洲梨、贴梗海棠和苹果的每同义位点同义替换数(Ks)分布。

图4 贴梗海棠中齐墩果酸和熊果酸的拟议生物合成途径及β-香树脂醇合酶基因簇。(A)八种植物中编码齐墩果酸和熊果酸生物合成途径中酶的基因数量。箭头上方每个框内的阿拉伯数字表示在苹果(Malus domestica)、杏(Prunus armeniaca)、桃(Prunus persica)、樱花(Prunus yedoensis)、白桦梨(Pyrus betulifolia)、欧洲梨(Pyrus communis)、玫瑰(Rosa chinensis)和贴梗海棠(C. speciosa)中编码该酶的基因家族的成员数量。(B)和(C)贴梗海棠第9号染色体上两个β-香树脂醇合酶基因簇的分布。
全基因组重复(WGD)和共线性分析
WGD是贴梗海棠基因组大小扩展的另一个主要事件。通过自共线性分析表征了贴梗海棠基因组中的共线性和WGD(图3C)。结果数据揭示了共线性染色体对,如LG01和LG07、LG02和LG15、LG03和LG11、LG04和LG12、LG05和LG10、LG06和LG14、LG08和LG15,以及LG09和LG17。这些结果表明贴梗海棠基因组中发生了WGD事件。为了理解WGD,选用了蔷薇科六个物种的基因组进行了染色体进化分析(图3D和E)。跨基因组共线性分析的结果揭示了玫瑰和杏之间潜在的染色体型式重排。这些重排的特征是杏的1号和8号染色体与玫瑰的3号染色体之间、杏的1号和3号染色体与玫瑰的5号染色体之间、杏的2号和6号染色体与玫瑰的6号染色体之间,以及杏的2号和7号染色体与玫瑰的7号染色体之间的共线性关系(图3E)。有趣的是,共线性分析得到的簇表明杏和桃之间存在1:1的共线性关系(图3D和E)。与此同时,结果中的共线性关系簇显示,欧洲梨、苹果和贴梗海棠位于同一个簇中,并在约4600万年前(MYA)发生分化。这一数据表明,这三个物种经历了四倍化事件,导致其染色体倍增至16条。为了进一步表征这些直系同源染色体和基因对,计算了这六个物种的每同义位点同义替换数(Ks)。结果数据在这些植物的Ks分布图上显示出一个共同的峰值,发生在4.2-4.3之间,表明这四个物种可能经历了一个双子叶植物特异的WGD事件(图3F)。此外,在贴梗海棠、苹果和欧洲梨的基因组中观察到Ks分布的一个显著峰值,发生在0.2-0.3之间(图3F)。这一结果表明,该事件可能与欧洲梨、贴梗海棠和苹果中最近发生的四倍化事件有关。
齐墩果酸和熊果酸的代谢途径构建
齐墩果酸和熊果酸是两种五环三萜类化合物,被中国药典指定为贴梗海棠的特征性化学标志物,具有药用价值。通过基因注释和序列挖掘,获得了参与贴梗海棠中这两种化合物生物合成的候选基因(图4A)。这些候选基因支持了从甲瓦龙酸途径到这两种化合物的生物合成步骤的构建(图4A)。为了表征与从牻牛儿基二磷酸开始的步骤特异性相关的基因,比较了贴梗海棠和其他七种物种中四个基因家族(鲨烯合酶、鲨烯2,3-氧化环化酶、香树脂醇合酶和β-香树脂醇28-单加氧酶)的大小(图4A)。结果数据确定了在贴梗海棠基因组中发现了11个β-香树脂醇合酶基因,其中10个分布在第9号染色体上的两个基因簇中。基因簇1的范围从12,157,281 bp到12,428,418 bp,包含8个β-香树脂醇合酶基因、9个其他基因和64个LTR逆转座子(图4B)。基因簇2的范围从31,206,028 bp到31,327,645 bp,包含2个β-香树脂醇合酶基因、5个其他基因和29个LTR逆转座子(图4C)。有趣的是,在所检查的基因组中,没有发现编码α-香树脂醇28-氧化酶的基因家族,这需要进一步的研究。对基因家族的表征进一步揭示了贴梗海棠基因组中的2个鲨烯合酶成员、12个鲨烯2,3-氧化环化酶成员、11个β-香树脂醇合酶成员和9个β-香树脂醇28-单加氧酶成员。与所检查的其他蔷薇科物种相比,贴梗海棠基因组中的鲨烯合酶、β-香树脂醇合酶和β-香树脂醇28-单加氧酶基因家族显著扩展,这与该物种中高含量的齐墩果酸和熊果酸相关。
讨论
我们的基因组测序和组装为促进贴梗海棠的遗传育种工作和栽培利用提供了宝贵的信息。贴梗海棠不仅是一种观赏作物,还是一种具有重要经济价值的药用和食用植物,在全球范围内广泛种植。然而,由于对其遗传学的了解有限,其遗传育种相对较慢。此外,木瓜属的五个物种在基因组、分子生物学等方面都未得到充分表征。长读长测序技术、组装算法和端粒到端粒(T2T)基因组已经被证明是组装水稻和西瓜等作物高质量基因组的优秀方法。在本研究中,我们使用这些技术为贴梗海棠开发了一个几乎无缺口的T2T基因组,结果显示其异质性较高。k-mer分析表明,贴梗海棠基因组的异质性率为2.1%,明显高于梨(0.89%)、苹果(0.85–1.28%)、桃(0.31%)、枇杷(0.31%)、梅(0.75%)、山楂(1.77%)和李子(0.92%)的基因组。此外,贴梗海棠的染色体级别基因组组装了632.3 Mb的序列和contig N50为32.3 Mb,对于未来的育种工作具有重要价值,使其复杂性可以与其他已经进行遗传育种测序的蔷薇科重要作物进行比较。我们的组装显示,贴梗海棠的基因组大小与苹果(652-668 Mb)相当,比山楂(779.24 Mb)和枇杷(733.32 Mb)小,但比梨(496.9-541.34 Mb)大。有趣的是,贴梗海棠基因组中发生了WGD事件,但在这些物种中并未发生。贴梗海棠中LTR的收缩含量低于苹果。更重要的是,最终的T2T基因组包括11条染色体两端具有端粒重复序列,四条染色体在单端具有端粒重复序列。因此,我们的T2T基因组为比较理解蔷薇科植物基因组特征提供了重要资源,因为T2T基因组的组装有助于理解端粒重复序列是否在其他植物物种中普遍存在。一方面,最近组装的苹果T2T基因组显示,七条染色体两端有端粒,八条染色体单端有端粒。一种野生型梨的T2T基因组组装显示,五条染色体的两端和八条染色体的单端也检测到了端粒重复序列。另一方面,在组装的山楂、枇杷和栽培梨的基因组中未发现端粒重复序列,这表明需要持续进行基因组组装,以理解蔷薇科植物的基因组进化。
与其他植物相比,贴梗海棠的T2T基因组组装揭示了12条拟染色体由单个contig表示,4条拟染色体由两个contig表示,1条拟染色体由四个contig表示。这些特征表明在贴梗海棠基因组的组装中存在七个间隙。初步分析表明,由于在间隙区存在大量的分散重复和倒位重复片段,这些间隙无法连续组装。此外,我们的T2T基因组以较高的contig N50、BUSCO、LAI和短读长比对率为特征。这些结果表明,本研究中组装的贴梗海棠基因组具有高度的连续性、完整性和准确性。基于基因组测序在提高苹果和其他作物经济价值方面的育种应用,可以预期,本研究中组装的基因组将对未来开发具有附加值的贴梗海棠新品种的育种工作具有重要意义。
直系同源聚类分析有助于识别与蔷薇科植物中不同生物或代谢过程相关的物种特异性基因家族。富集分析揭示了枇杷的物种特异性基因家族,这些基因家族与基因组重组和修复的独特遗传和分子机制的进化有关。另一项富集分析显示,山楂的物种特异性基因家族参与了生物合成和代谢过程,如淀粉和蔗糖代谢及脂肪酸降解。梨的谱系特异性基因家族与DNA代谢过程、DNA整合、DNA重组和纤维素微原纤维组织有关。我们的富集分析显示,贴梗海棠的多个特异性基因家族主要参与代谢过程,如半乳糖代谢和萜类骨架生物合成。此外,对Hap1和Hap2的特异性基因进行保守结构域富集分析,表明它们富集到一些与逆转录酶相关的保守结构域,表明这些特异性基因源自逆转座子的转位。我们的组装估计了贴梗海棠基因组中扩展或收缩的基因家族。这种扩展和收缩在蔷薇科植物中基因的功能多样化中起着重要作用。据报道,山楂基因组中扩展的基因家族参与了植物天然产物的生物合成途径。在枇杷基因组中,发现扩展的基因家族与单萜类生物合成和淀粉及蔗糖代谢有关。我们的基因功能注释发现,贴梗海棠基因组中扩展的基因家族数量几乎是收缩的两倍。这些扩展的基因家族参与了单萜类生物合成、黄酮类生物合成、倍半萜类和三萜类生物合成等。β-香树脂醇合酶在贴梗海棠中比其他已测序的蔷薇科植物中更为冗余,这表明LTR介导了基因组中此类酶的近端复制,并在第9号染色体上形成了两个β-香树脂醇合酶基因簇。通过分析其他基因组,我们发现玫瑰在第2号染色体上有一个包含5个β-香树脂醇合酶的基因簇。有趣的是,迄今为止研究的所有其他蔷薇科植物中都没有β-香树脂醇合酶基因簇,尽管它们的这一基因家族中有两个到四个成员。
基于这些结果,我们进一步研究了参与贴梗海棠果实中健康促进三萜类化合物生物合成的基因家族。贴梗海棠中齐墩果酸和熊果酸生物合成的主要途径基因家族得到了鉴定。尽管这些基因家族在贴梗海棠中的功能需要进一步研究,但这些基因家族的注释,例如苹果中的α-香树脂醇合酶和β-香树脂醇合酶酶的注释,支持了我们的功能注释。此外,贴梗海棠基因组中两个基因家族的扩展程度高于蔷薇科其他植物。这些数据支持了该物种中齐墩果酸和熊果酸的高产量,因此对于进一步阐明贴梗海棠中植物次级代谢具有重要价值。
结论
我们为贴梗海棠组装了一个T2T和染色体级别的基因组。这是第一个来自蔷薇科木瓜属的植物基因组。这个高质量的基因组具有较高的异质性,大小为650.4 Mb。该基因组锚定在17条拟染色体上,contig N50为35.5 Mb,支架N50为35.8 Mb。十二条拟染色体由单个contig表示,组装中存在七个间隙。十一条拟染色体在两端有端粒重复序列,四条拟染色体在单端有端粒重复序列。此外,对贴梗海棠基因组中所有拟染色体的着丝粒区域进行了预测。共注释了45,515个编码蛋白质的基因。还识别了扩展或收缩的物种特异性基因家族。功能注释表明,主要扩展的基因家族与植物次级代谢有关,从而能够构建药用齐墩果酸和熊果酸的生物合成途径。LTR介导了贴梗海棠基因组中β-香树脂醇合酶基因的多次近端复制,与其他已测序的蔷薇科植物相比,该物种中β-香树脂醇合酶基因的成员更多。组装的高质量基因组为研究贴梗海棠中药用化合物的生物合成和代谢工程提供了宝贵的平台。
材料与方法
植物材料
贴梗海棠(C. speciosa)是一种二倍体灌木,在中国湖北省武汉市的校园研究站内培育了大量用于研究的植株。健康的叶片、茎、根、花和果实被收集后,迅速用液氮冷冻,并储存在−80°C的冰箱中,直至使用。
高保真测序与基因组组装
基因组DNA从叶片中提取,使用Plant Genomic DNA kit(Tiangen,北京,中国)进行高保真(HiFi)测序,这是第三代测序技术。使用SMRTbell Express Template Prep Kit 2.0(Pacific Biosciences of California,Menlo Park,CA,USA)构建单分子实时(SMRT)测序文库,并在PacBio Sequel II系统上使用一个8 M SMRT细胞进行测序。为了获得HiFi读数,使用ccs软件(GitHub - PacificBiosciences/ccs: CCS: Generate Highly Accurate Single-Molecule Consensus Reads (HiFi Reads))处理子读数。基因组大小、异质性和重复序列比例通过jellyfish(GitHub - gmarcais/Jellyfish: A fast multi-threaded k-mer counter)和GCE(GitHub - fanagislab/GCE: GCE (genomic charactor estimator) is a bayes model based method to estimate the genome size, genomic repeat content and the heterozygsis rate of the sequencing sample. The estimated result can be used to design the sequencing strategy.)的K-mer分析(k=21)估计。然后,使用Hifiasm在默认的haplotig清除参数和‘–primary’的设置下,将高质量的HiFi读数组装成contig。此外,使用参数‘Hi-C-partition: -hl/2’进行了单倍体组装,并添加了Hi-C测序数据进行单倍体组装和分型。
单倍体变异的检测
为了检测单倍体SNP-InDel,使用nucmer(—mum, —maxgap=500, —mincluster=100)进行共线性比对,并使用delta-filter(−1, −q, −r)程序过滤结果。然后,使用show-snps输入SNP和InDel位点,这些程序是MUMmer(v4.0)的子程序。为了将SNP和InDel位点文件转换为vcf格式文件,使用MUMmerSNPs2VCF.py脚本。使用MummandCo(V3.0)在默认参数下检测两个单倍体之间的SV,并使用GenomeSyn-1.2.7绘制两个单倍体的共线性图。
染色体构象捕获测序与支架组装
染色体构象捕获(Hi-C)测序方法用于在染色质消化后生成150 bp的双端读长。使用FastQC(www.bioinformatics.babraham.ac.uk/projects/fastqc/)评估读长质量,并使用fastp进行过滤。使用HiCUP(www.bioinformatics.babraham.ac.uk/projects/hicup/)鉴定有效的读长对,并使用BWA将其比对到contig上。使用Juicer 1.6生成染色体接触频率图,并使用3D-DNA流程(GitHub - aidenlab/3d-dna: 3D de novo assembly (3D DNA) pipeline)进行支架组装。使用Juicebox Assembly Tools(GitHub - aidenlab/Juicebox: Visualization and analysis software for Hi-C data -)手动校正支架。使用BUSCO分析评估基因组组装的完整性。使用D-GENIES将支架比对到苹果参考基因组,以确定其身份。
Iso-seq测序
从不同植物组织中提取RNA样本,使用TRIzol试剂(Invitrogen,Carlsbad,CA,USA)。使用SMARTer™ PCR cDNA合成试剂盒(Takara Biotechnology,大连,辽宁,中国)合成cDNA。使用Pacific Biosciences DNA Template Prep Kit 2.0构建SMRTbell文库,并由Frasergen Bioinformatics Co., Ltd(Frasergen,武汉,湖北,中国)在PacBio Sequel II平台上进行测序。
着丝粒和端粒的鉴定
通过检查端粒区域中是否存在重复序列(TTAGG)来进行端粒序列的鉴定。使用Centromics17程序,以基因组序列数据、原始HiFi测序数据和Hi-C测序数据为输入进行着丝粒鉴定。
重复元件的鉴定
使用Tandem Repeats Finder在默认参数下识别串联重复序列并进行软掩蔽,同时使用BEDTools。通过同源性搜索和ab initio预测进行重复分析。使用RepeatMasker(www.repeatmasker.org)基于RepBase(v.21.12)库(www.girinst.org/repbase)检测同源序列,并根据先前报道的协议使用LTR_Finder识别LTR-RT。使用RepeatModeler(www.repeatmasker.org/RepeatModeler/)构建ab initio预测重复库。结合LTR和de novo重复库,使用RepeatMasker筛选基因组组装。通过从EDTA输出文件计算LTR组装指数分数来评估组装的完整性。
基因预测和功能注释
为了预测编码蛋白质的基因,使用了三种方法:同源搜索、de novo预测和Iso-seq数据。在重复掩蔽的基因组上使用BRAKER2流程预测基因模型,并基于NCBI、TrEMBL、InterPro和Swiss-Prot蛋白质数据库中的非冗余蛋白质序列(nr)和BLASTP以及KEGG数据库对其功能进行注释,E值阈值为<1E-5。使用PfamScan和InterProScan注释蛋白质结构域,并使用PFAM数据库识别基因模型中的基序和结构域。使用Blast2GO注释基因本体(Gene Ontology)ID。
此外,使用各种工具注释非编码RNA基因。tRNA通过tRNAscan-SE(GitHub - UCSC-LoweLab/tRNAscan-SE: A program for detection of tRNA genes)使用真核参数进行预测,而mRNA、snRNA和snoRNA则通过INFERNAL(Infernal: inference of RNA alignments)基于Rfam和miRbase数据库进行预测。rRNA及其亚基通过RNAmmer(http://cbs.dtu.dk/services/RNAmmer/)进行预测。
基因家族鉴定和系统发育分析
为了构建系统发育树,使用了11个物种的基因组:贴梗海棠、苹果、杏、桃、樱花、白桦梨、欧洲梨、玫瑰、番木瓜、葡萄和拟南芥(用作外群)。NCBI提供了每个物种的全基因组蛋白质序列下载。使用OrthoFinder构建11个物种的基因家族,并识别单拷贝直系同源组以建立系统发育树。使用MAFFT对每个单拷贝直系同源组进行氨基酸比对,而核苷酸比对则使用PAL2NAL创建,网址为www.bork.embl.de/pal2nal/。然后,基于所有单拷贝基因的拼接比对,使用IQ-TREE建立最大似然系统发育树。使用PAML中的MCMCTREE评估物种分歧时间。最后,使用CAFE探讨基因家族的扩展和收缩。
全基因组重复分析
为了分析WGD事件,选择了贴梗海棠、苹果、欧洲梨、杏、玫瑰和桃的基因组。通过计算基因组中的每同义位点同义替换数(Ks)确定WGD事件。使用BlastP识别每个基因组中的同源物,e值的阈值为<1e−5。使用ParaAT对同源物进行编码蛋白质DNA比对(ParaAT - Tools - BIG Data Center - National Genomics Data Center (CNCB - NGDC))。然后使用KaKs_Calculator计算Ks值(KaKs - BIG Data Center - National Genomics Data Center (CNCB - NGDC))。为了绘制Ks分布图,我们使用了R的ggplot2包。
这篇关于贴梗海棠T2T基因组-文献精读40的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!