fastq专题

如何从BAM文件中提取fastq
虽然高通量测序分析最常用的操作是将fastq比对到参考基因组得到BAM文件,但偶尔我们也需要提取BAM文件中特定区域中fastq。最开始我认为这是一个非常简单的操作,因为samtools其实已经提供了相应的工具samtools fastq. 以biostar handbook的Ebola病毒数据为例,首先获取比对得到的BAM文件。 # 建立文件夹mkdir -p refs# 根据Acces

如何使用fastq-dump转换SRA格式
如何使用fastq-dump转换SRA格式 做生信的基本上都跟NCBI-SRA打过交道,尤其是fastq-dump大家肯定不陌生.NCBI的fastq-dump软件一直被大家归为目前网上文档做的最差的软件之一”,而我用默认参数到现在基本也没有出现过什么问题,感觉好像也没有啥问题, 直到今天看到如下内容, 并且用谷歌搜索的时候,才觉得大家对fastq-dump的评价非常很到位. 我们一般使用

全外显子测序分析流程1 - Fastq质控与去接头、低质量和引物序列
全外显子测序分析流程1 - Fastq质控与去接头、低质量和引物序列 1. 运行实例 # -d 样本根目录# -s 样本名称python trim_fastq.py -d /result/WES/sample -s sample 2. fastqc质控报告与去接头、低质量序列主程序 对raw fastq和clean fastq生成质控QC报告trim_galore去接头、低质量序列和
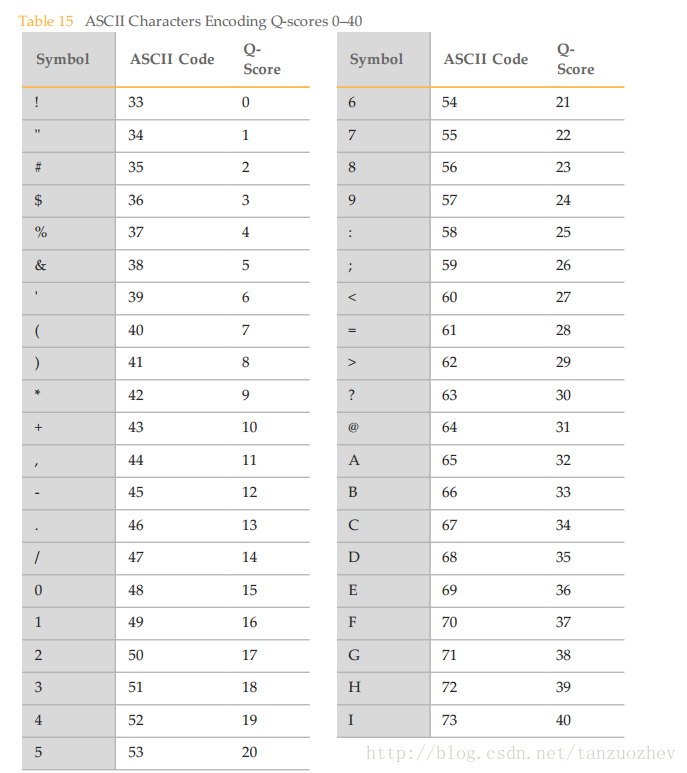
Illumina Fastq Q-score
Illumina Nextseq500 Miseq HiseqXten 测序仪 Q-score均采用下面的编码格式,仅作简要介绍。 Q-score Q-score 在fastq中每个序列的第4行,代表测序错误的概率。 Quality Score Q(X) ## Error Probability P(~X) Q40 ## 0.0001 (1 in 10,000) Q30 ## 0.0

python 随机抽取Fastq文件
参考 http://pythonforbiologists.com/index.php/randomly-sampling-reads-from-a-fastq-file/ 最近要做一个二代测序的模拟,所以网上找了个小脚本,做了些注释,希望能够帮助大家。 from __future__ import divisionimport randomnumber_to_sample = 300000
绘生信技能41 - Matplotlib绘制测序Fastq数据ATCG碱基分布图
输入文件数据 sample_id sample_name cycle_num A_ratio C_ratio G_ratio T_ratio N_ratio Q24B2807-01 XXZ 1 0.229 0.334 0.3 0.123 0.014 Q24B2807-01 XXZ 2 0.251 0.32 0.254 0.157 0.018 Q24B2807-01 XXZ 3 0.296 0.2
转录组学习第三弹-下载SRR数据并转成fastq
下载数据 前面已经安装好了需要的软件,那么我们现在需要下载我们练习需要用到的sra数据。从 SRA 数据库下载数据有多种方法。可以用ascp快速的来下载 sra 文件,也可以用wget或curl等传统命令从 FTP 服务器上下载 sra 文件。另外sra-tools的prefetch也支持直接下载。在此处我用的是prefetch,因为ascp我尝试了很多次都没成功,遂放弃了。 说明:由于数据量

三代ONT测序的fast5和fastq数据中pass、fail和skip三个文件夹含义
牛津纳米孔技术(ONT,Oxford Nanopore Technologies)公司的生产的测序产品从小到大,常用的主要是接512通道芯片的MinION和接3000通道芯片的PromethION两款测序仪。其中两者均可生产包括原始fast5格式数据(包含测序条件等数据)和碱基识别后的fastq格式数据(含质量值的数据)。如图最新一次MinION下机数据,其中电脑驱动软件为MinKNOW(内置Gu